
Abstract
Depression is the most common psychiatric disorder of the developed world with a lifetime prevalence of between 7.5 and 17% [1,2]. Originally conceived by Kraepelin to be a non-deteriorating, recurrent disorder with remissions, the discovery of effective pharmacotherapy in the 1950s provided hope for patients. Despite the extensive use of anti-depressants, however, two long-term studies of patients with depression suggested a different picture and depression is increasingly being viewed as a chronic, disabling condition [3,4].
Since the discovery of antidepressant medications, over 40 years of research has been conducted into the aetiology and psychopharmacology of depression. Yet, despite this, two fundamental problems in antidepressant treatments remain. First, up to 30% of patients with depression will not respond to the first choice of antidepressants administered, and while 10–15% may be helped by a change of class of antidepressant or by electroconvulsive therapy (ECT), a small group remain chronically ill and resistant to treatment [5]. Second, all antidepressant regimens are associated with a latency period of 1–2 weeks before there is noticeable clinical effect, despite rapid blockade of monoamine uptake or catabolism.
One solution hypothesised to improve both the latency of clinical effect and the problem of treatment resistance has been the addition of the beta blocker pindolol for its additional 5-hydroxytryptamine (5HT1A) antagonist properties. This paper reviews the clinical effectiveness of pindolol in combination with antidepressant treatments, the pharmacodynamic rationale for its use and, finally, we propose an alternative pharmacokinetic hypothesis yet to be tested.
Clinical studies in pindolol/antidepressant combinations
A number of clinical studies have reported the use of 5HT1A antagonist pindolol in combination with antidepressants following promising preclinical work utilising in vivo microdialysis and electrophysiological techniques. It was hoped that by blocking the proposed negative serotonergic feedback (see pharmacodynamic rationale) there would be improvements in latency of clinical response and augmentation of antidepressant effect in depression. Studies in patients with depression have included both open-label and placebo-controlled, double-blind strategies.
Open-label studies
The first open-label study of patients with depression using pindolol/antidepressant combinations was reported by Artigas et al. [6] in 1994 and was followed by a study by Blier et al. [7] in 1995. Both were small studies of similar design that included two separate parts to examine (i) latency of clinical response and (ii) augmentation in treatment resistant cases (see Tables 1 and 2). Of the seven patients in the study of Artigas et al., four achieved a remission within 1 week of combination treatment, while Blier et al. reported seven of nine patients remitting within 1 week of addition of pindolol [7]. In the ‘treatment resistant’ parts of the studies, remission was reported in five of eight and 10 of 19 patients, respectively [6,7]. While all patients in the latency parts of both studies received paroxetine (20 mg/day), the treatment-resistant patients were continued on a number of different classes of antidepressant including monoamine oxidase inhibitors (MAOIs), tricyclic antidepressants (TCAs) and specific serotonin reuptake inhibitors (SSRIs), affording the opportunity to compare class differences. Although the numbers were small, MAOIs appeared to show reduced response and while those on paroxetine and fluoxetine had high remission rates, none of five patients taking sertraline achieved remission [6,7].
Pindolol/antidepressant combinations: open trials, unspecified patients
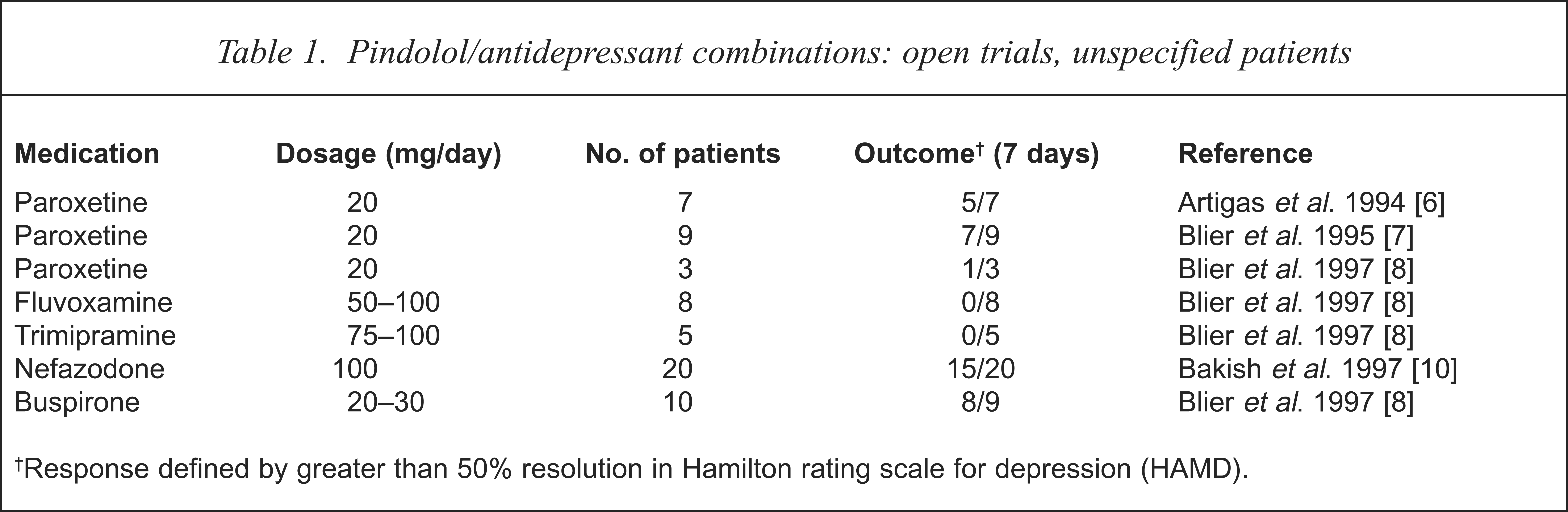
Response defined by greater than 50% resolution in Hamilton rating scale for depression (HAMD).
Pindolol/antidepressant combinations: open trials, ‘treatment resistant’ patients

Response defined by greater than 50% reduction in Hamilton rating scale for depression (HAMD) except for
only mean HAMD reported.
Three subsequent open-label studies have since reported on pindolol/antidepressant combinations in patients with depression. Blier et al. attempted to replicate their earlier study with pindolol in combination with SSRIs (paroxetine, fluvoxamine), TCAs, (desipramine, trimipramine) and the 5HT1A agonist (buspirone), in non-resistant patients [8]. While response to paroxetine was less robust (one of three patients achieved remission within 1 week) none of the eight patients treated with fluvoxamine and none of the five taking trimipramine responded at 1 week.
Dinan et al. reported much lower response rates in a study comparing intraclass differences in SSRI/pindolol combinations in treatment-resistant patients with a single responder for each group of patients taking fluoxetine (six patients), paroxetine (three patients) and sertraline (four patients) [9]. The failure in response with sertraline was consistent with the Blier et al. [7] study; however, the lower response rates with paroxetine and fluoxetine were unexpected. The final open-label study reviewed the efficacy of pindolol/nefazodone combinations and reported 40% remission rate at 1 week and 90% at 2 weeks [10].
Although there are some encouraging results among the open-label studies, there is a variety of general and specific weaknesses worthy of discussion. The general issue of inherent bias in open-label studies has been well discussed in the literature [11]. The studies reviewed employed small numbers and inadequate statistical methods, and did not account for the placebo response, which is known to be up to 25% in augmentation studies [12]. It is unclear from the studies how much of the effect could be accounted for by increased compliance, closer contact with researchers and the possibility that beta blockade by pindolol reduced depression rating scores through the blocking of peripheral manifestations of anxiety.
More specific issues relate to the ‘treatment resistant’ groups in the Artigas et al. [6] and Blier et al. [7] studies and the assessment of latency of response. The definition of ‘treatment resistance’ is unstated and, in fact, many of the patients would not be considered ‘resistant’ by current definitions [13]. The adequacy of previous trials was insufficiently explored, and in the Blier et al. [7] study the duration of current treatment was unspecified and five patients had received only a single trial of antidepressant. In assessing latency of response, Artigas et al. [6] asked the patients to contact the investigator ‘as soon as they started to feel better’ and they were then examined on the following days. This is not an accepted method of establishing early response. Patients in a novel trial might be expected to focus on early improvements, and there is no indication in the Artigas et al. [6] study of whether improvements were maintained; however, this is more clearly established in the Blier et al. [7] study which had a 28-day follow-up.
Placebo-controlled, double-blind studies
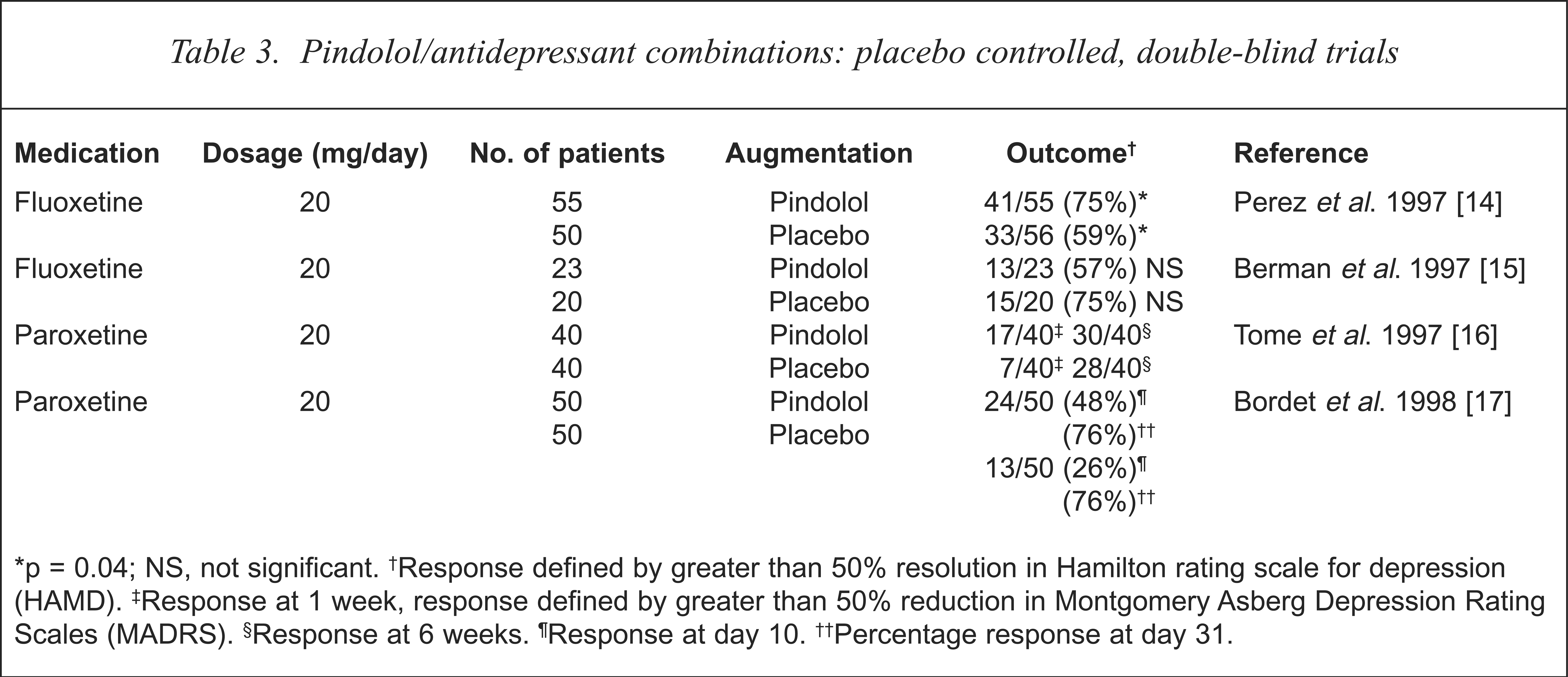
Several placebo-controlled, double-blind studies have been reported which improve on previous open-label studies (see Table 3). Following a 7-day placebo run-in phase, Perez et al. randomly assigned 111 patients with depression (DSM-IV) receiving fluoxetine to either pindolol (7.5 mg/day) or placebo [14]. Those taking pindolol/fluoxetine combinations showed a greater number of responders (45/55 vs 33/56, p = 0.04), greater number of sustained responders (33/55 vs 27/56, p = 0.03) and greater falls on depression rating scales, (Hamilton rating scale for depression (HAMD) 13.5 vs 11.2, p = 0.04). The placebo-controlled, double-blind trial of Berman et al. [15], on the other hand, found different results in 43 patients treated with paroxetine (20 mg/day) randomised to either pindolol (7.5 or 10 mg/day) or placebo. Using a number of clinician-rated and self-rated scales, they were unable to detect any statistical differences between groups on depression rating, within a group by depression rating, or group effects on ceasing pindolol (apart from Beck depression inventory scores, p = 0.05). In contrast to Blier et al. [7], but consistent with Perez et al. [14], Berman et al. [15] detected significant clinical signs of beta blockade.
Pindolol/antidepressant combinations: placebo controlled, double-blind trials
p = 0.04; NS, not significant.
Response defined by greater than 50% resolution in Hamilton rating scale for depression (HAMD).
Response at 1 week, response defined by greater than 50% reduction in Montgomery Asberg Depression Rating Scales (MADRS).
Response at 6 weeks.
Response at day 10.
Percentage response at day 31.
The third major study examining the efficacy of pindolol/SSRI combinations is that of Tome et al. [16], who randomised patients on paroxetine (20 mg/day) to either pindolol (7.5 mg/day) or placebo in a 6-week study. The study was performed at two centres, at the first centre there were no significant differences at day 14, while at the second centre, pindolol-treated patients had significant reductions in mean scores on the Montgomery Asberg Depression Rating Scale (MADRS). Those at the second centre had better prognostic signs, such as increased general practitioner referral rate and increased employment, while those at the first centre, who did not show significantly increased response, had higher rates of previous episodes of depression and use of psychotropics. After 6 weeks of treatment, pindolol administration was ceased and improvements remained, which Tome et al. [16] interpreted as representing possible long-lasting effects of pindolol treatment but which may also suggest significant group differences at the two centres.
While both Perez et al. [14] and Tome et al. [16] claimed improved efficacy with the addition of pindolol to SSRI treatments, neither of their studies showed reduced latency of response. Bordet et al. found the opposite [17]. In a placebo-controlled, double-blind study of 100 patients with depression who were treated with paroxetine (20 mg/day) and pindolol (15 mg/day) or placebo and rated at days 5, 10, 15, 21, 25, 31, 60, 120 and 180, the workers reported significant between-group differences of remission at day 10 but not days 5 or 15 and thereafter. There were statistically significant differences in mean HAMD scores at days 5 and 10 but in clinical terms these were small, amounting to about 3 points on the HAMD (19.0 vs 15.7; 14.7 vs 11.7, respectively). While the methodology of this study conformed to most of the minimum requirements of speed on onset studies laid down by Prien et al. [18], the frequency of clinical measures was insufficient and the statistical analyses unsophisticated in determining differences in latency of effect.
The only other placebo-controlled, double-blind study of pindolol compared pindolol with fluoxetine (or placebo) augmentation of subtherapeutic doses of trazodone (100 mg/day) in treatment-resistant patients (as defined by the criteria of Thase and Rush [13]) [19]. Both pindolol or fluoxetine augmentation groups showed statistically significant improvements in response compared with placebo (p = 0.006) at 5 weeks.
Unfortunately, the placebo-controlled, double-blind studies do not sufficiently clarify the value of adding pindolol to antidepressant regimens in terms of improved latency or efficacy in depression. The only controlled study attempting to clarify augmentation of efficacy in treatment-resistant cases is marred by inadequate pre-trial doses of antidepressant, small numbers (eight to 10 patients per group) and subtherapeutic doses of trazodone [19]. In testing the improved latency hypothesis, few placebo-controlled studies have sufficiently frequent clinical measures to distinguish early onset or response, and those that did reported conflicting results. In the three studies of sufficient sample size and placebo-controlled double-blind methodology, there are conflicting results as to whether pindolol provides improved efficacy when added to SSRI regimens [14–16].
The pharmacodynamic rationale for 5HT1A antagonists/antidepressant combinations
There is good evidence that facilitation of 5HT neurotransmission may act either directly or indirectly as a unifying mechanism of antidepressant treatments [20]. One of the major conundrums in the pharmacotherapy of depression, however, has been the delay in response of 1 to 2 weeks, despite immediate uptake blockade/reduced metabolism of monoamines at a synaptic level.
Three major theories have been advanced to explain this delay: (i) timing of post-synaptic events including receptor down-regulation and cellular changes subsequent to G-protein activity and nuclear changes; (ii) feedback mechanisms of monoamines onto presynaptic autoreceptors; and (iii) pharmacokinetic mechanisms. The pharmacodynamic rationale for the use of 5HT1A antagonists involves the second of these three mechanisms.
The 5HT1A receptors are a sub-group of G-protein-coupled 5HT receptors which exist post-synaptically and in presynaptic locations on the soma and dendrites of 5HT neurones. While the somatodendritic 5HT1A receptors are found mainly in the mid- and dorsal raphe nuclei of the mid-brain, post-synaptic 5HT1A receptors are found in limbic projections of serotonergic neurones (e.g. frontal cortex, hippocampus) [21]. The presynaptic somatodendritic 5HT1A receptors have an autoinhibitory function and, when stimulated by 5HT, result in decreased electrical activity (firing), 5HT synthesis and 5HT release [22–24] (see Figure 1).
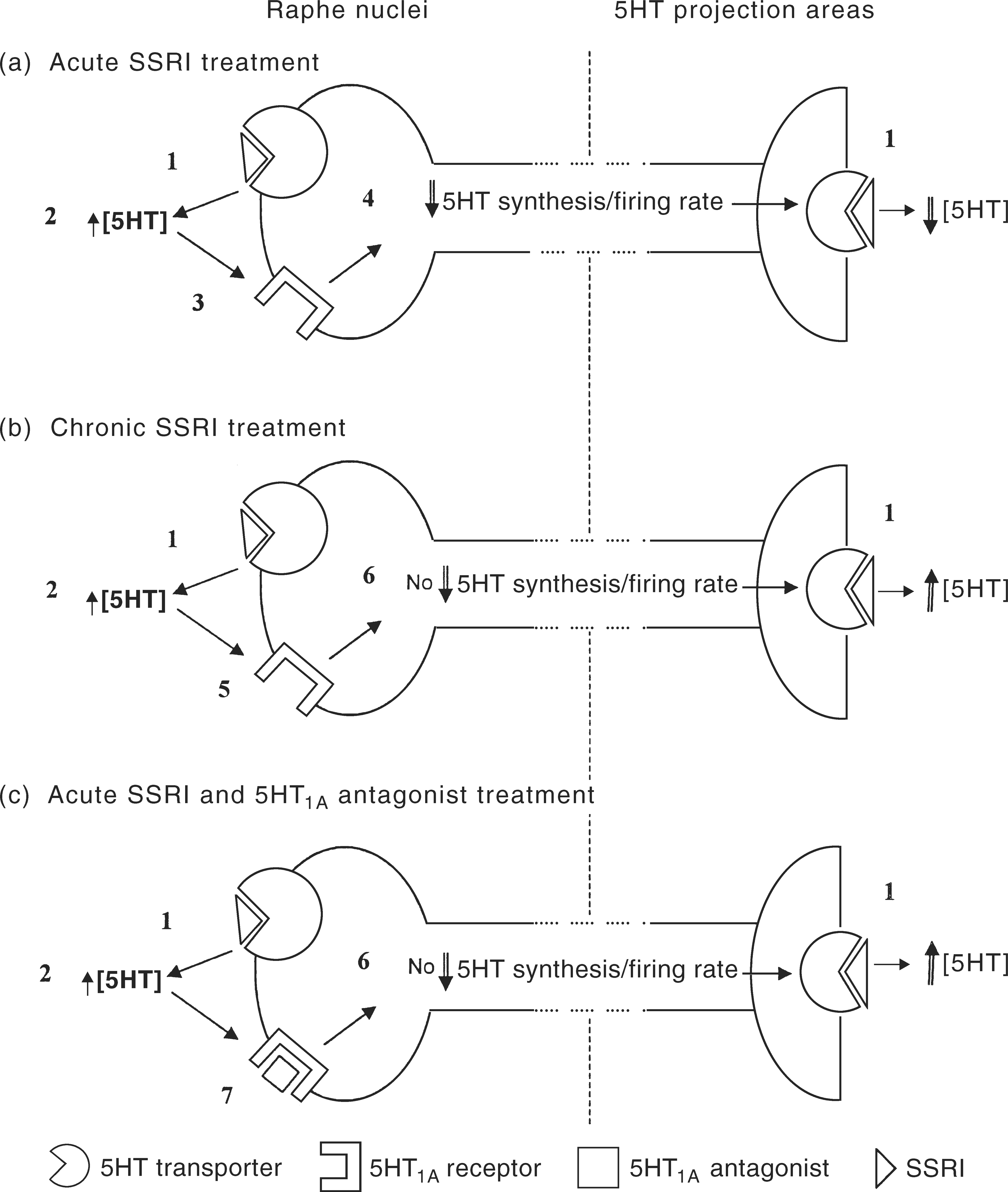
The Pharmacodynamic Rationale for 5HT1A Antagonist Augmentation of SSRI's. In acute SSRI administration, (a) there is uptake blockade of somatodendritic and synaptic sites (1). The resultant elevations of extracellular 5HT (2) feedback onto somatodendritic 5HT1A receptors (3) which leads to reductions of 5HT synthesis and neuronal firing rate and synaptic 5HT concentrations. Following repeated SSRI administration (b), there is a down regulation of somatodendritic 5HT1A receptors (5) which reduces negative feedback autoregulation and there is a normalisation of 5HT synthesis and neuronal firing (6) leading to net elevations of synaptic 5HT concentration. The addition of 5HT1A antagonists to acute SSRI administration (c) blocks initial 5HT negative feedback onto somatodendritic 5HT1A receptors, (7) thus preventing initial reductions in 5HT synthesis and neuronal firing rate and results in early net increases in synaptic 5HT concentrations. An early antidepressant response may then be expected consequent on downregulation of postsynaptic 5HT receptors.
Electrophysiological studies in rats have shown that repeated administration of SSRIs (and other antidepressants) leads to a functional desensitisation of somatodendritic 5HT1A receptors, the timing of which correlates well with the delay in onset of action of SSRIs [20, 25]. In effect, the initial rise in extracellular 5HT following SSRI administration feeds back onto 5HT1A somatodendritic receptors in raphe nuclei, resulting in inhibition of the neurone, reducing firing and reduced 5HT synthesis. With repeated administration of SSRIs, there is a down-regulation of somatodendritic 5HT1A receptors, reduced negative feedback autoinhibition and increased firing and 5HT synthesis. When this occurs in the presence of on-going 5HT uptake blockade at the synapse by SSRIs, there is a facilitation of 5HT neurotransmission and only then an antidepressant response (see Figure 1).
The rationale for administering 5HT1A antagonists in combination with SSRIs involves a functional blockade of initial 5HT feedback onto somatodendritic 5HT1A receptors. With somatodendritic 5HT1A receptors effectively blocked, the 5HT neurone continues to fire and synthesise 5HT while SSRIs increase synaptic 5HT concentrations through uptake-pump blockade. Theoretically, this may result in earlier antidepressant response through agonist-induced post-synaptic down-regulation of 5HT receptors.
Preclinical studies of rats, using microdialysis techniques, have shown that administration of both selective (WAY 100635) and non-selective (pindolol) 5HT1A antagonists, in combination with SSRIs, enhances extracellular 5HT in various projections of the 5HT neurones, including frontal cortex, hypothalamus, striatum and hippocampus [26–30]. Pindolol alone, however, has little effect on 5HT concentrations. It is unlikely that this effect in humans can be accounted for by presynaptic 5HTID blockade (for which pindolol has much lower affinity) [31] or β-adrenergic effects as metoprolol, a beta blocker which lacks 5HT1A antagonism, does not potentiate increases in extracellular 5HT in combination with SSRIs such as citalopram [32].
A pharmacokinetic rationale
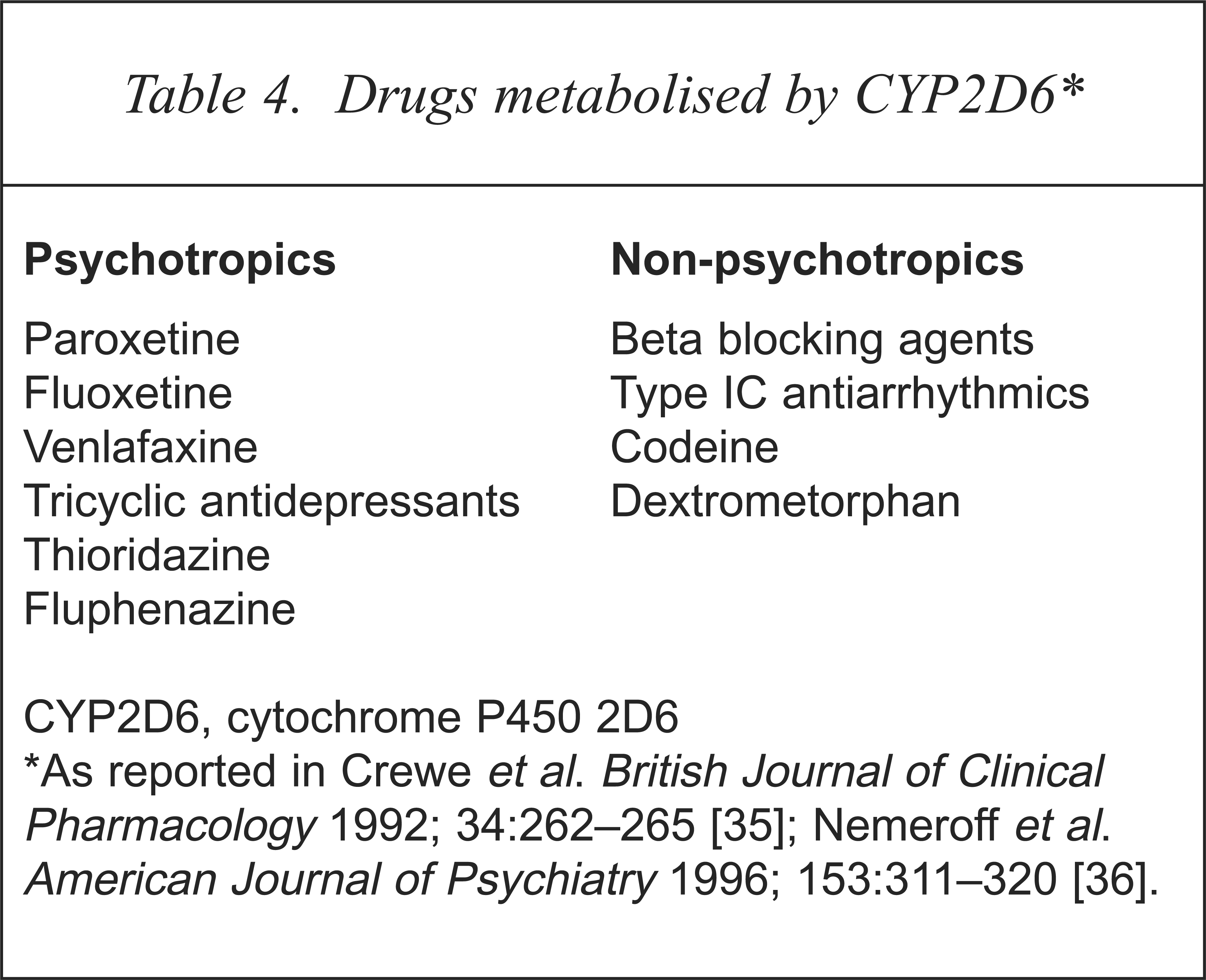
Much of the theoretical rationale for the putative benefits of 5HT1A antagonists/antidepressant combinations is centred around the pharmacodynamic mechanism, while there has been little investigation into the possible role of pharmacokinetic interactions in this strategy. Many drugs, including the SSRIs, undergo extensive hepatic biotransformation by the cytochrome P450 enzyme system [33]. While cytochrome P450 2D6 (CYP2D6) is the most extensively studied of the cytochromes, the CYP450 system includes more than 30 related haemoprotein enzymes located both in hepatic and in extrahepatic sites and participates in the oxidation of a wide variety of psychotropic and other drugs [34].
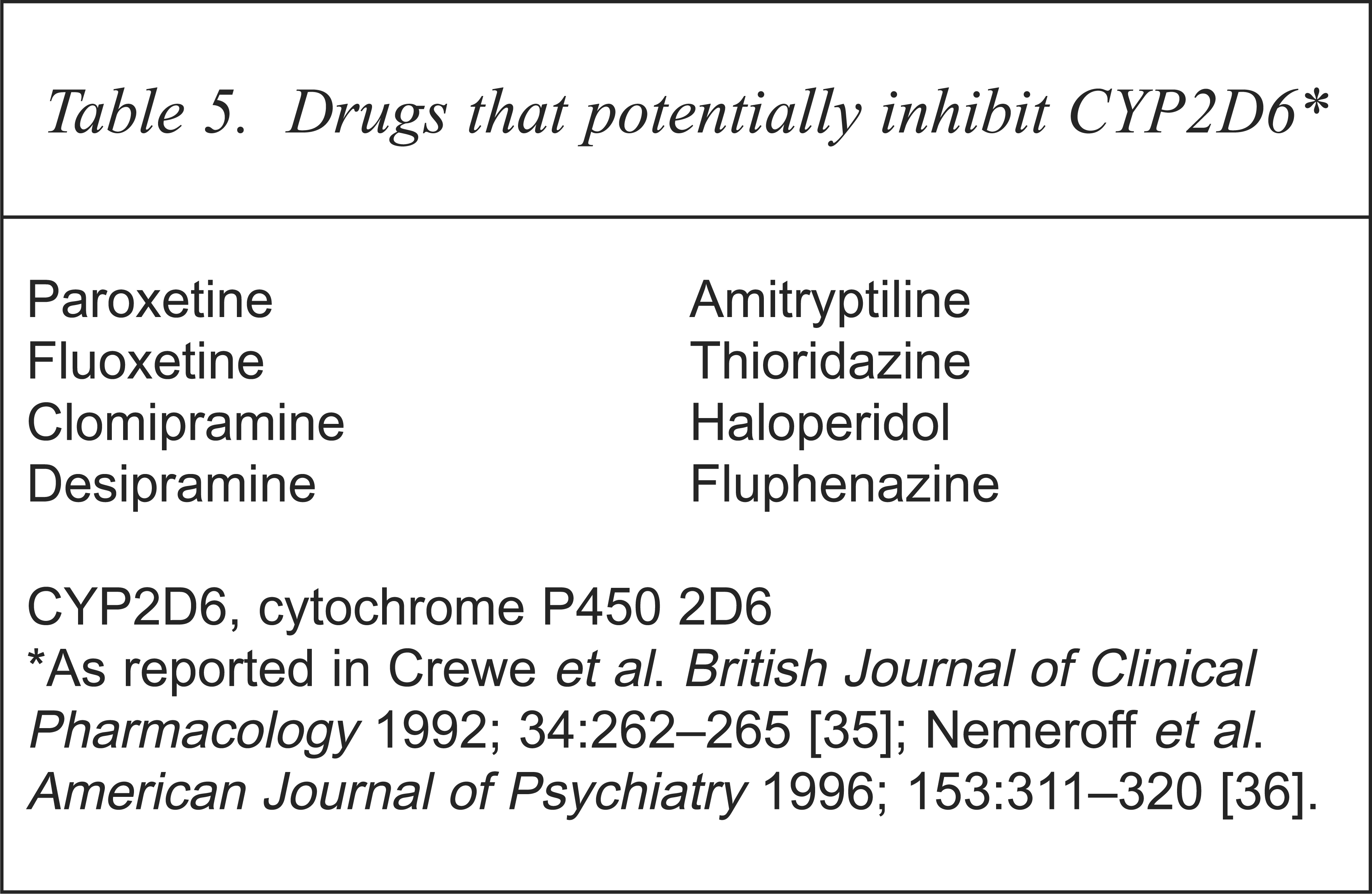
The cytochrome CYP2D6 has been implicated in the biotransformation of a number of psychotropic and non-psychotropic agents (Table 4) [35,36]. In addition, a number of these drugs, including the SSRIs paroxetine and fluoxetine, potently inhibit CYP2D6 (Table 5) [34,35]. Competitive drug–drug interactions are therefore highly likely at this site and these may result in non-linear pharmacokinetics. Indeed, case reports have shown elevations of plasma levels of nortriptyline, perphenazine, haloperidol and clozapine in combination with fluoxetine [36–39], and both fluoxetine (20 mg/day) and paroxetine (20 mg/day) have led to one- to three-fold increases in the plasma levels of desipramine [33,37,39]. Given that paroxetine and fluoxetine both are metabolised by and inhibit CYP2D6, and that the beta blockers, including pindolol, also have high affinity for CYP2D6 and may inhibit it, there exists a high possibility of competitive drug–drug interactions, resulting in elevated plasma levels, with these combinations. While this has not been formally studied in large groups, three- to four-fold increase in paroxetine levels following the introduction of pindolol in a patient with depression has been reported [40].
Drugs metabolised by CYP2D6∗
CYP2D6, cytochrome P450 2D6
Drugs that potentially inhibit CYP2D6∗
CYP2D6, cytochrome P450 2D6
Reports of clinical success with pindolol augmentation primarily involve paroxetine and fluoxetine, two agents that inhibit CYP2D6. On the other hand, sertraline, which does not significantly inhibit CYP2D6, has not had similar clinical reports of success with pindolol augmentation. The question remains, does increasing the plasma level of an SSRI increase the response in treatment-resistant depression or speed the onset of therapeutic effect?
In support of the notion that greater and faster responses may be achieved by exposing central receptor sites to higher drug levels in the initial phases of treatment, it has been reported that pulse-loading doses of imipramine lead to rapid and complete antidepressant response [41]. While this was a small study, it was methodologically sound, using a double-blind placebo-controlled design. Intravenous pulse-loading with clomipramine has been shown to lead to rapid response both in patients with depression [42] and in obsessive–compulsive disorder (OCD) patients [43] in placebo-controlled, double-blind trials.
Thus, these small trials support the idea that early, elevated plasma levels of drugs (and presumably drug levels acting at central receptor sites) can hasten the onset of action. The possibility exists that increasing drug levels through a pharmacokinetic interaction, such as by the addition of pindolol, may achieve the same result as does simply increasing the dose. Clearly, further controlled studies are necessary to test this hypothesis.
Conclusion
Preclinical studies provide a plausible pharmacodynamic rationale for the addition of 5HT1A antagonists to antidepressant regimens in the treatment of depression. While there is some evidence that such augmentation strategies in open-label studies improve latency of clinical response and efficacy in treatment-resistant cases, the improved methodology of large placebo-controlled, double-blind studies have produced conflicting results. The strongest support for pindolol augmentation is seen in combinations with fluoxetine or paroxetine, but the apparent failure of augmentation with sertraline cannot be explained by pharmacodynamic mechanisms. While there are as yet few studies, there is a strong possibility of drug–drug interactions when pindolol is added to antidepressant regimens that rely on CYP2D6 metabolism. The resultant elevations in antidepressant drug levels may provide an alternative hypothesis for the success of pindolol augmentation strategies, though further studies are required to test this. Pindolol is not ideal for augmentation because of its broad-spectrum pharmacodynamics. While a number of selective 5HT1A antagonists may become available for future clinical use, the current absence of selective compounds, the conflicting clinical results of 5HT1A antagonist augmentation and the potentially fatal consequences of pindolol overdose means that pindolol augmentation of antidepressants should be considered an experimental approach.