
Abstract
The main objective of this study, on mice, was to compare the neuroprotective effects of propofol with those of propofol plus disodium edetate (propofol EDTA). We also administered propofol EDTA (0.005% (w/v) EDTA) to mice intravenously, and measured the changes in zinc concentrations occurring after permanent middle cerebral artery occlusion. In the in vivo study, propofol EDTA displayed stronger neuroprotective effects than propofol alone. Furthermore, we examined the neuroprotective effects of EDTA administered alone, and found that EDTA Na significantly reduced the infarct volume. The number of terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling-positive cells in the ischemic penumbra was reduced more by propofol EDTA than by propofol alone. We performed in the in vitro study in five groups (aerobic, vehicle (control), propofol, EDTA, and propofol plus EDTA). Propofol and EDTA each protected PC12 cells against oxygen—glucose deprivation-induced cell damage, and the effect of propofol was increased by adding EDTA. Because the chelating action of EDTA was a potential causal mechanism, we examined the effect of propofol EDTA on intracerebral zinc homeostasis. When propofol EDTA was given intravenously 10 mins before cerebral ischemia, the zinc concentration decreased significantly in the cortical area, but not in the subcortex. In conclusion, (a) propofol provides neuroprotection against both in vivo and in vitro ischemic damage, and its effects are enhanced when EDTA is added; and (b) EDTA itself protects against ischemic neuronal damage, possibly, owing to its zinc-chelating action.
Introduction
Propofol (2,6-diisopropylphenol) is a potent intravenous hypnotic agent, and is widely used both for the induction and maintenance of anesthesia as well as for sedation in the intensive care unit (Marik, 2004). Laboratory investigations have revealed that propofol might also protect the brain against ischemic injury. Numerous characteristics of propofol have been reported: (a) it reduces cerebral blood flow, the cerebral metabolic rate for oxygen, and intracranial pressure (Pittman et al, 1997); (b) it acts as an antioxidant, directly scavenges free radicals, and decreases lipid peroxidation (Sagara et al, 1999; Wilson and Gelb, 2002); (c) it activates γ-aminobutyric acid type A receptors (Ito et al, 1999), inhibits glutamate receptors (Zhan et al, 2001), and reduces the extracellular glutamate concentration via an inhibition of Na+ channel-dependent glutamate release or an enhancement of glutamate uptake (Sitar et al, 1999); and (d) it reportedly reduces ischemic neuronal injury in animal models of transient global and focal cerebral ischemia (Young et al, 1997), although conflicting results have been obtained in studies carried out using models of permanent middle cerebral artery occlusion (MCAO). Indeed, although Tsai et al (1994) failed to show a protective action of propofol after permanent MCAO in the same model, Adembri et al (2006) reported that it reduced infarct size.
This study was therefore designed to evaluate the neuroprotective properties of propofol, primarily by determining whether it alters the volume of the infarct resulting from permanent focal cerebral ischemia in mice. For the following reasons, we also (a) asked whether a modulating influence over the neuroprotective effects of propofol might be exerted by disodium edetate (EDTA) and (b) tested the effects of EDTA on intracerebral zinc concentrations during the cerebral ischemia. The physiologic significance of neuronal zinc release within the central nervous system is not clear, moreover, its role in ischemic brain injury is controversial (Shabanzadeh et al, 2004). Indeed, although Miyawaki et al (2004) concluded that the synaptic release of Zn2+, which is potently neurotoxic and its translocation into postsynaptic neurons might be implicated in ischemic cell death, other authors (Matsushita et al, 1996) stress that zinc may have a protective function. EDTA, which is usually included in pharmaceutical preparation of propofol to retard bacterial and fungal growth, has the ability to chelate almost every positive ion in the periodic table. We hypothesized that chelation of excessive neuronal zinc might ameliorate zinc-induced neurotoxicity and reduce subsequent neuronal injury. We therefore compared the neuroprotective effects of propofol EDTA and propofol alone. Because the presence of EDTA enhanced the neuroprotective effects of propofol, determined of the intracerebral zinc concentrations to know whether EDTA exerted this modulating effect by chelating surplus intracerebral zinc.
Materials and methods
Animals
The experiments were conducted in accordance with the Animal Care Guidelines issued by the Animal Experiments Committee of Gifu Pharmaceutical University. All efforts were made to minimize both suffering and the number of animals used. All in vivo experiments were performed using male ddY mice (5-week old; body weight 26 to 32 g; Japan SLC Ltd., Shizuoka, Japan). The animals were housed at 24 ± 2°C under a 12 h light—dark cycle (lights on from 0700 to 1900). Each animal was used for one experiment only.
Drugs
For this study, 1% Diprivan® injection and 1% Propofol injection ‘Maruishi’ were purchased from AstraZeneca Co. Ltd. (Osaka, Japan) and Maruishi Co. Ltd. (Osaka, Japan), respectively. Intralipid and EDTA Na were from Terumo Co. Ltd. (Tokyo, Japan) and Sigma (Deisenhofen, Germany), respectively. Pentobarbital and isoflurane were from Nissan Kagaku (Tokyo, Japan) and Merck Hoei Ltd. (Osaka, Japan), respectively. 2,3,5-Triphenyltetrazolium chloride (TTC), ethylenediaminetetraacetic acid (EDTA) disodium salt dihydrate, Dulbecco's modified Eagle's medium, and resazurin were all from Sigma-Aldrich Co. (St Louis, MO, USA). Nitric acid for atomic absorption spectrometry and hydrogen peroxide for atomic absorption spectrochemical analysis were purchased from Kanto Chemical Co. Inc. (Tokyo, Japan) and Wako Pure Chemical Industries (Osaka, Japan), respectively. Hoechst 33342 and YO-PRO-1 were from Molecular Probes (Eugene, OR, USA).
Focal Cerebral Ischemia Model in Mice
Anesthesia was induced using 2.0 to 3.0% isoflurane and maintained using 1.0 to 1.5% isoflurane in 70% N2O/30% O2 by means of an animal general anesthesia machine (Soft Lander, Sin-ei Industry Co. Ltd., Saitama, Japan). Body temperature was maintained at 37.0 to 37.5°C with the aid of a heating pad and heating lamp. In each mouse, regional cerebral blood flow was monitored by laser-Doppler flowmetry (Omegaflow flo-N1; Omegawave Inc., Tokyo, Japan). A flexible probe was fixed to the skull (2 mm posterior and 6 mm lateral to bregma). After a midline skin incision, the left external carotid artery was exposed, and its branches were occluded (Hara et al, 1996, 1997). An 8–0 nylon monofilament (Ethicon, Somerville, NJ, USA) coated with a mixture of silicone resin (Xantopren; Bayer Dental, Osaka, Japan) was introduced into the left internal carotid artery through the external carotid artery stump so as to occlude the origin of the middle cerebral artery (MCA). Afterwards, the left common carotid artery was occluded. After the surgery, the mice were kept in a cage with a heating lamp, which maintained the cage temperature between 29 and 30°C for another 3 h. Then, the mice were bred in a preoperative condition (24 ± 2°C) until sampling.
Propofol Treatment
Control (vehicle), propofol EDTA, and propofol groups received 10% intralipid, 1% Diprivan® injection (which includes 0.005% (w/v) disodium edetate (EDTA)), and 1% Propofol injection ‘Maruishi’, respectively. Mice were randomly assigned to one of the three groups to receive propofol with EDTA, propofol alone, or vehicle intravenously (0.1 ml/10 g, over 90 secs) 10 mins before MCAO. Propofol (with or without EDTA) was administered at 1, 5, or 10 mg/kg (total seven groups). In an experiment to examine time dependency, propofol (10 mg/kg, with or without EDTA) or intralipid were administered 10 mins before, after, or 1 h after MCAO (total nine groups). In addition, we prepared another three were groups (intralipid (vehicle), propofol EDTA, and propofol) to evaluate the survival rate, infarct volume, and neurologic deficits at 7 days after MCAO. Mice received propofol (with or without EDTA, 10 mg/kg, intravenously) or intralipid intravenously 10 mins before MCAO.
EDTA Na Treatment
Mice were randomly assigned to receive EDTA Na (0.05 or 0.5 mg/kg) or vehicle intravenously (0.1 ml/10 g, over 90 secs) 10 mins before MCAO.
Physiologic Monitoring
A polyethylene catheter inserted into the left femoral artery was used to measure arterial blood pressure and heart rate (Power Lab/8SP; AD Instrument, Osaka, Japan) 20 mins before and 30 mins after MCAO. Propofol (with or without EDTA) was administered intravenously (10 mg/kg, over 90 secs, 0.1 ml/10 g) at 10 mins before MCAO. Blood samples (50 μL) were taken before and at 30 mins after the onset of ischemia for pharmacokinetic analysis. pH, pCO2, pO2, and glucose were measured (i-STAT 300F, Abbot Co., Abbot Park, IL, USA). Regional cerebral blood flow was measured during this period by the method described in the section Focal cerebral ischemia model in mice. Physiologic monitoring was carried out a separate study.
Assessment of Cerebral Infarct and Cerebral Edema Volumes
To analyze infarct volume, mice were euthanized using sodium pentobarbital at 24 h or 7 day after MCAO, and forebrains were sectioned coronally into five slices (2 mm thick) and placed in 2% TTC at 37°C for 30 mins. The infarcted areas were recorded as images using a digital camera (Coolpix 4500, Nikon, Tokyo, Japan), then quantitated (using an Image J), and calculated as in a previous report (Hara et al, 1997). Brain swelling was calculated according to the following formula: (infarct volume + ipsilateral undamaged volume—contralateral volume) × 100/contralateral volume (%) (Hara et al, 1996).
Neurologic Deficits
Mice were tested for neurologic deficits at 24 h or 7 day after MCAO and scored as described in our previous study (Hara et al, 1996) using the following scale: 0, no observable neurologic deficits (normal); 1, failure to extend the right forepaw (mild); 2, circling to the contralateral side (moderate); 3, loss of walking or righting reflex (severe). The investigator who rated the mice was masked as to the group to which each mouse belonged.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End-Labeling Staining
The terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling staining (TUNEL) assay was performed according to the manufacturer's instructions (Roche Molecular Biochemicals Inc., Mannheim, Germany). Ischemic areas of cortical brain sections of 0.4 to 1.0 mm anterior to bregma (through the anterior commissure) were excised and used. For this part of the study, we referred to a mouse brain atlas (George Paxinos and Keith BJ Franklin, The Mouse Brain in Stereotaxic Coordinates, 2nd edition, 2001). Paraffin-embedded sections were dewaxed and rehydrated, and then incubated in 20 mg/ml of proteinase K for 30 mins. After immersion in 100 μml of 3% H2O2 for 30 mins, sections were incubated in a terminal deoxynucleotidyl transferase (TdT) labeling reaction mixture (supplied with the kit) in a humidified chamber for 90 mins at 37°C, then incubated in the stop buffer at 37°C for 4 mins.
Cell Counting
To quantify the number of DNA-fragmented cells present after MCAO, the numbers of TUNEL-positive cells in the caudate-putamen (as the ischemic core) and cortex (as the ischemic penumbra; two areas, the superior and inferior cortical areas) were counted in a high-powered field (×200) on a section through the anterior commissure by a masked investigator. Each count was expressed as number/mm2 (n = 6 or 7).
Measurement of Intracerebral Zinc Concentrations
Forty mice were assigned to the following experimental groups (10 mice per group): (1) vehicle (no ischemia) group; (2) propofol EDTA (no ischemia) group; (3) vehicle (ischemia) group; and (4) propofol EDTA (ischemia) group. Propofol EDTA (10 mg/kg) or vehicle was administered intravenously (0.1 ml/10 g, over 90 secs) 10 mins before MCAO, and mice were euthanized using sodium pentobarbital at 1 h after MCAO. Non-ischemic groups were killed 1 h after administration of drug or vehicle. The resected brain was promptly divided into cortex and subcortex under a microscope. The areas perfused by the anterior cerebral artery and posterior cerebral artery were then removed from the cortexes. The resected samples were incubated at 100°C for 24 h to remove edema, and dry samples were weighed. The concentration of zinc present in each tissue was determined using the following method. Two milliliters of nitric acid was added to each dried sample followed by gradual heating from 70 to 140°C. The following day, 0.5 ml of nitric acid and 2 ml of H2O2 were added to each sample, followed (as before) by gradual heating from 70 to 140°C. The dry samples were brought to 4.0 ml with deionized water, and the zinc concentration in the samples was determined using ICP-MS (HP 4500; Yokokawa Analytical Systems, Tokyo, Japan). Mass numbers of m/z 64 and 66 m/z were used for these determinations.
Cell Culture
PC12 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated horse serum and 5% heat-inactivated fetal bovine serum at 37°C under a humidified 5% CO2 atmosphere. To examine the effect of propofol on oxygen—glucose deprivation (OGD)-induced cell death, cells were seeded into collagen-coated 24-well plates at a density of 1 × 104 cells/well. After incubating for 7 days, the culture medium was removed and the cells, after being washed twice with glucose-free Dulbecco's modified Eagle's medium, were incubated in the same glucose-free medium for 4 h in an oxygen-free incubator (94% N2, 5% CO2, and 1% O2). After OGD, sufficient glucose was added to bring it to the normal level (final concentration, 4.5 mg/ml) and cells were incubated under normal growth conditions for an additional 18 h (reoxygenation). Propofol (10 or 20 μmol/L) or EDTA (0.05 μmol/L) was added to the glucose-free medium before OGD treatment.
Cell Viability
The first method used here for assessing cell viability was a single-cell digital imaging-based method employing fluorescent staining of nuclei. Cell death was assessed on the basis of combination staining with fluorescent dyes (namely, Hoechst 33342 and YO-PRO-1 (Molecular Probes)), observations being made using an inverted epifluorescence microscope (Olympus, Tokyo, Japan). YO-PRO-1 (λex 491 nm and λem > 509 nm), a membrane-impermeant dye, is generally excluded from viable cells, whereas early-stage apoptotic and necrotic cells are YO-PRO-1-positive. At the end of the culture period, Hoechst 33342 and YO-PRO-1 dyes were added to the culture medium (at 8 and 0.1 μmol/L, respectively) for 30 mins. Images were collected using a digital camera (Coolpix 4500). In a blind manner, a total of at least 400 cells per condition were counted using image-processing software (Image-J ver. 1.33f; National Institutes of Health, Bethesda, MD, USA). Cell mortality was quantified by determining the percentage of cells that were YO-PRO-1-positive (Hoechst 33342-positive cells being taken as the total number of cells present, as Hoechst 33342 stains are both live and dead cells).
Second, we examined the change in fluorescence intensity after cellular reduction of resazurin to resorufin. All experiments were performed in Dulbecco's modified Eagle's medium at 37°C. Cell viability was assessed after immersion in 10% resazurin solution for 3 h at 37°C, and fluorescence was recorded at 560/590 nm.
Statistical Analysis
All data are presented as mean ± s.d. Statistical comparisons were made using a one- or two-way analysis of variance followed by Student's t-test, Dunnett's test, Bonferroni correction, or the Mann—Whitney U-test. A χ2 test was used to identify differences in the survival rates. StatView software version 5.0 (SAS Institute Inc., Cary, NC, USA) was used. *P < 0.05 was considered statistically significant.
Results
Physiologic Parameters
Physiologic parameters are shown for all groups in Table 1. There were no significant differences in regional cerebral blood flow, mean arterial blood pressure, or heart rate, or in arterial pH, pCO2, pO2, or glucose among the vehicle, propofol, and propofol with EDTA groups. Surface regional cerebral blood flow was reduced to 19 to 22% of baseline regional cerebral blood flow immediately after MCAO in all mice. Glucose was increased to 142 to 143% of preischemic value after MCAO in all mice.
Physical parameters before and after drug administration
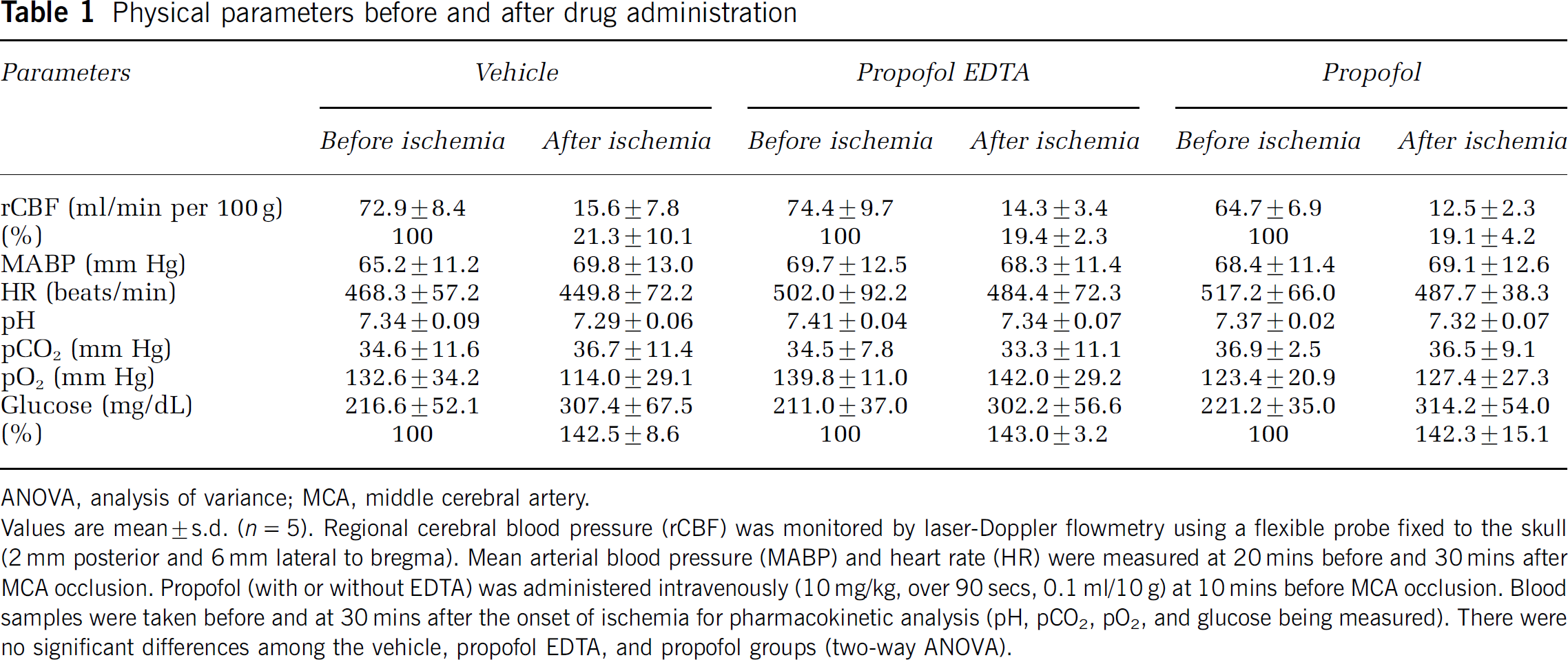
ANOVA, analysis of variance; MCA, middle cerebral artery.
Values are mean ± s.d. (n = 5). Regional cerebral blood pressure (rCBF) was monitored by laser-Doppler flowmetry using a flexible probe fixed to the skull (2 mm posterior and 6 mm lateral to bregma). Mean arterial blood pressure (MABP) and heart rate (HR) were measured at 20 mins before and 30 mins after MCA occlusion. Propofol (with or without EDTA) was administered intravenously (10 mg/kg, over 90 secs, 0.1 ml/10 g) at 10 mins before MCA occlusion. Blood samples were taken before and at 30 mins after the onset of ischemia for pharmacokinetic analysis (pH, pCO2, pO2, and glucose being measured). There were no significant differences among the vehicle, propofol EDTA, and propofol groups (two-way ANOVA).
Infarction, Edema, and Neurologic Deficits
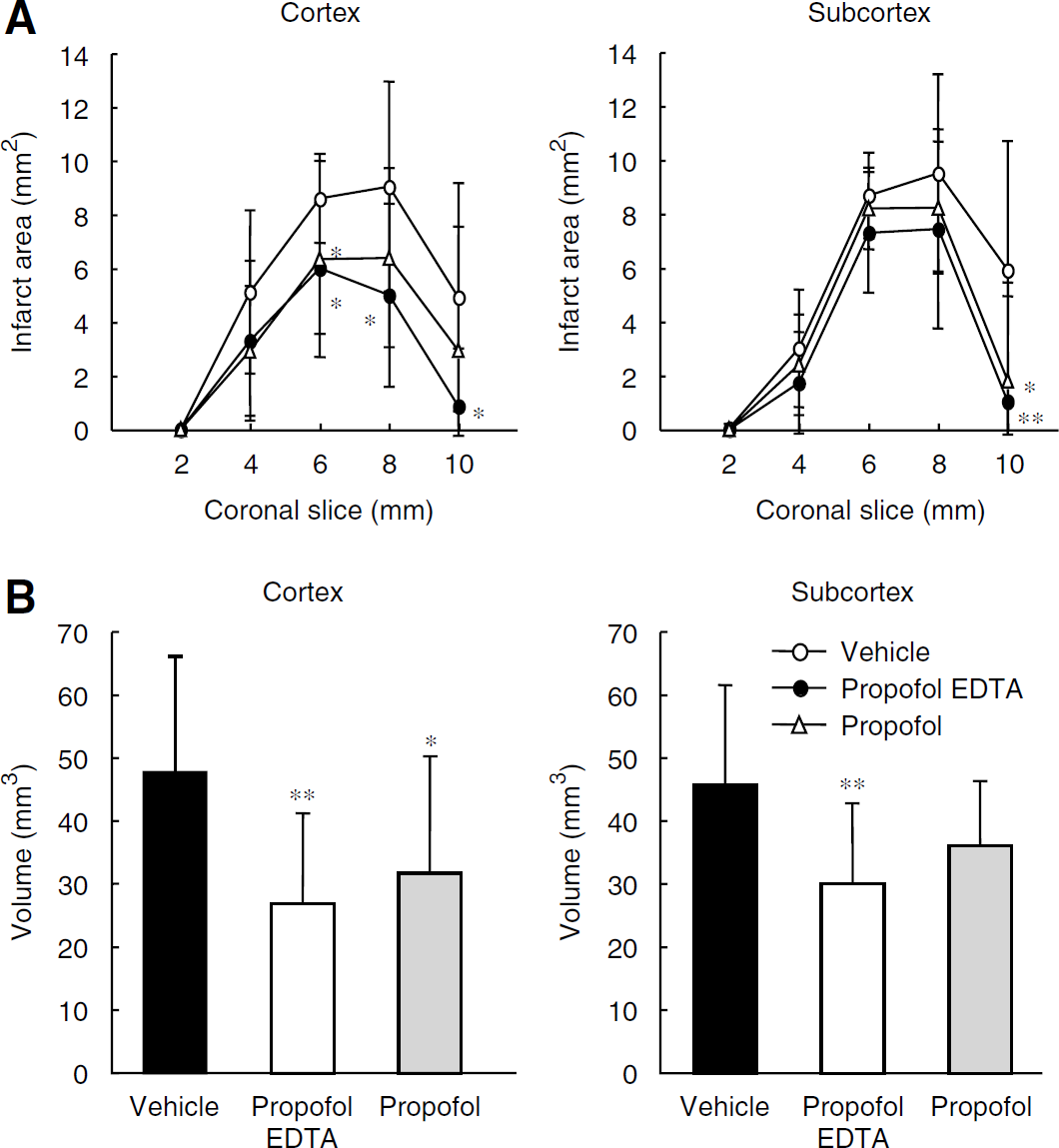
Using TTC staining, we examined whether propofol EDTA and propofol would reduce infarct volume. Twenty-four hours after MCAO, the mice had developed infarcts affecting the cortex and striatum. Mice that died within 24 h after the operation were excluded from the present experiment. Five of 42 (11.9% mortality), five of 64 (7.8%), and seven of 63 (11.1%) mice died in the intralipid, propofol EDTA, and propofol treatment groups, respectively. No significant differences in mortality rate were observed among the treatment groups. First, each drug preparation was administered intravenously 10 mins before MCAO. Administration of 5 or 10 mg/kg propofol EDTA or propofol (but not 1 mg/kg) significantly reduced infarct area, infarct volume, and brain swelling (Figures 1A to 1C), with propofol EDTA reducing infarct area and brain swelling significantly more than propofol. Similarly, propofol EDTA or propofol administered at 5 or 10 mg/kg (intravenously at 10 mins before ischemia), but not at 1 mg/kg, apparently reduced the neurologic deficits, although only propofol EDTA reduced them significantly (Figure 1D).
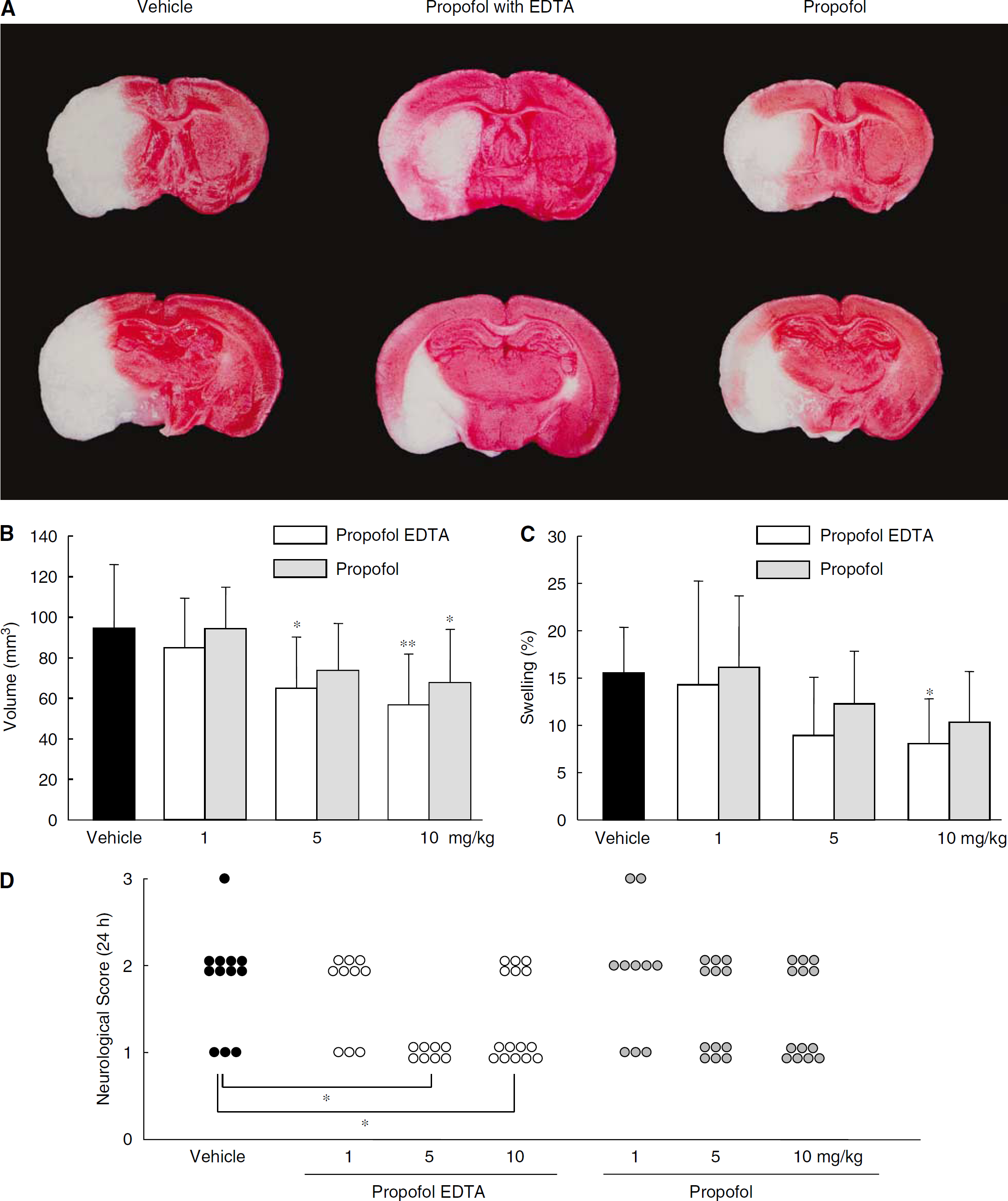
(
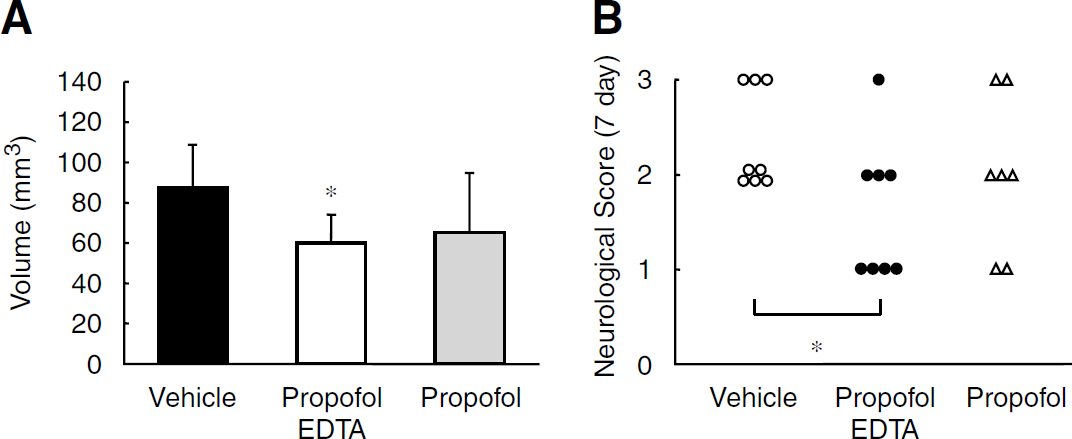
Next, we reviewed the time dependency of the drug efficacy of propofol EDTA and propofol. Propofol EDTA and propofol (10 mg/kg intravenously at 10 mins before or after MCAO) apparently decreased the cerebral infarction, whereas no clear effects were observed in the groups treated at 1 h after MCAO. Indeed, propofol EDTA and propofol, when administered 10 mins before MCAO, each significantly reduced infarct volume and brain swelling (Figures 2A and 2B). When propofol EDTA was administered after MCAO, infarct area and infarct volume were significantly reduced, but brain swelling was not (versus vehicle-treated mice). When administered after MCAO, propofol alone also displayed a neuroprotective effect, but the effect, was if any, weaker than that of propofol EDTA. When propofol EDTA or propofol was administered 1 h after MCAO, there was no reduction in infarct area, infarct volume, and brain swelling (edema) (versus vehicle-treated mice). The mortality rate, infarct volume, and neurologic deficit scores after MCAO were compared among the treatment groups when examined over a 7-day period. Seven of 15 (46.7% mortality), 6 of 13 (46.2%), and 7 of 14 (50.0%) mice died in the intralipid, 10 mg/kg propofol EDTA, and 10 mg/kg propofol treatment groups, respectively. No significant differences in mortality rate were observed among the treatment groups. Moreover, propofol EDTA decreased infarct volume significantly at 7 days after MCAO (versus vehicle) (Figure 3A). Thus, overall propofol EDTA displayed greater neuroprotection than propofol.
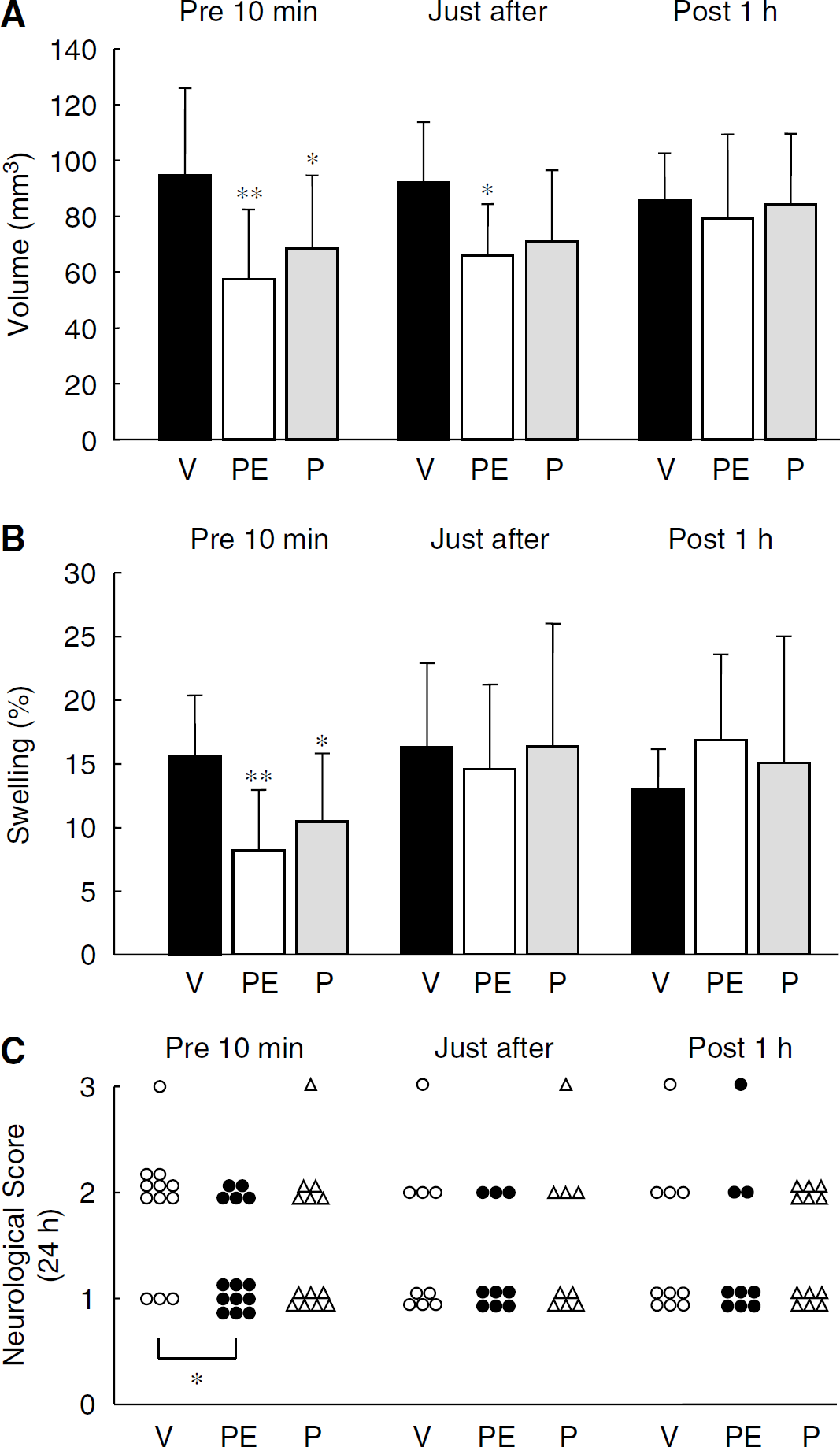
(
(
Similarly, administration of propofol EDTA at 10 mins before (but not at, after, or 1 h after) MCAO led to a significant reduction in the scores given for neurologic deficits (Figure 2C). In addition propofol EDTA decreased the neurologic scores significantly at 7 days after MCAO (versus vehicle) (Figure 3B). Further, regional analysis revealed that treatment with propofol EDTA or propofol at 10 mins before ischemia reduced both infarct area and infarct volume in the cortex (Figure 4), although only propofol EDTA reduced infarct volume in the subcortex.
(
Histologic Observation of Cerebral Ischemic Damage
The morphologic features of TUNEL-stained cells (representing the ischemic damage and apoptotic cell death induced by 12 or 24 h MCAO) are shown in Figure 5. Cells exhibiting shrunken cell bodies and condensed nuclei were distributed in both the ischemic core and penumbra of the territory affected by MCAO, with the TUNEL-positive cells being among the population displaying such features. In this study, TUNEL-positive cells were predominantly located in the ischemic core region rather than in the ischemic penumbra, and propofol EDTA significantly reduced the number of TUNEL-positive cells in the ischemic penumbra (Figures 5C and 5D). We next distinguished apoptotic cells from necrotic cells, and each type was counted. Only densely labeled cells showing cell shrinkage, chromatin condensation, and fragmented nuclei indicating apoptosis were considered to be apoptotic cells, whereas cells with light diffus e labeling were taken as necrotic (Figure 5B). We compared only the numbers of apoptotic cells (yellow bar, Figures 5C and 5D). Propofol EDTA reduced this count significantly in the ischemic penumbra area at 24 h (but not 12 h) after MCAO.
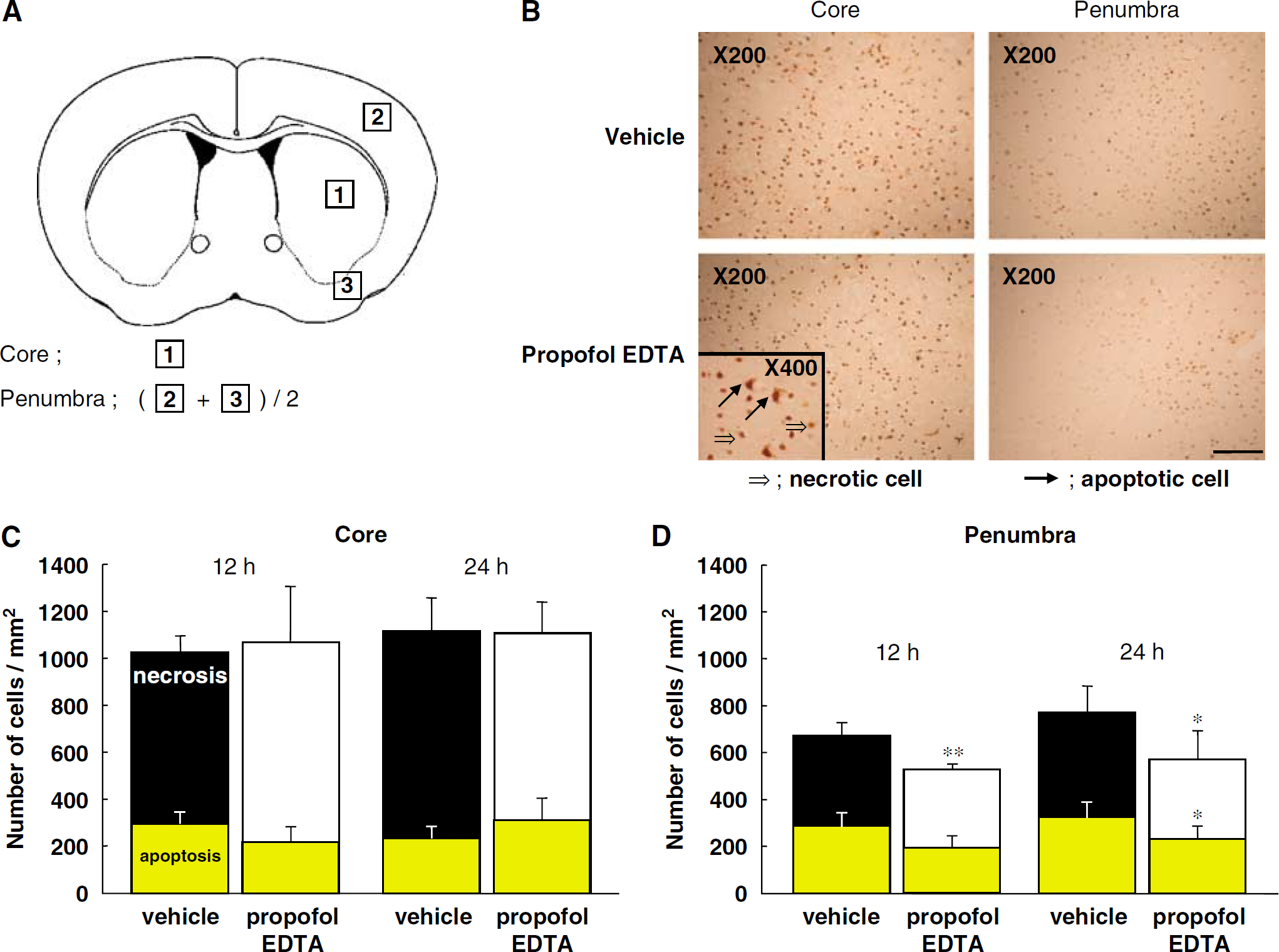
Effect of propofol EDTA on TUNEL staining after MCAO. Propofol EDTA (10 mg/kg intravenously) was administered 10 mins before MCAO, and mice were killed at 12 or 24 h after the occlusion. Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling staining was performed as described in Materials and methods. (
EDTA Na Treatment
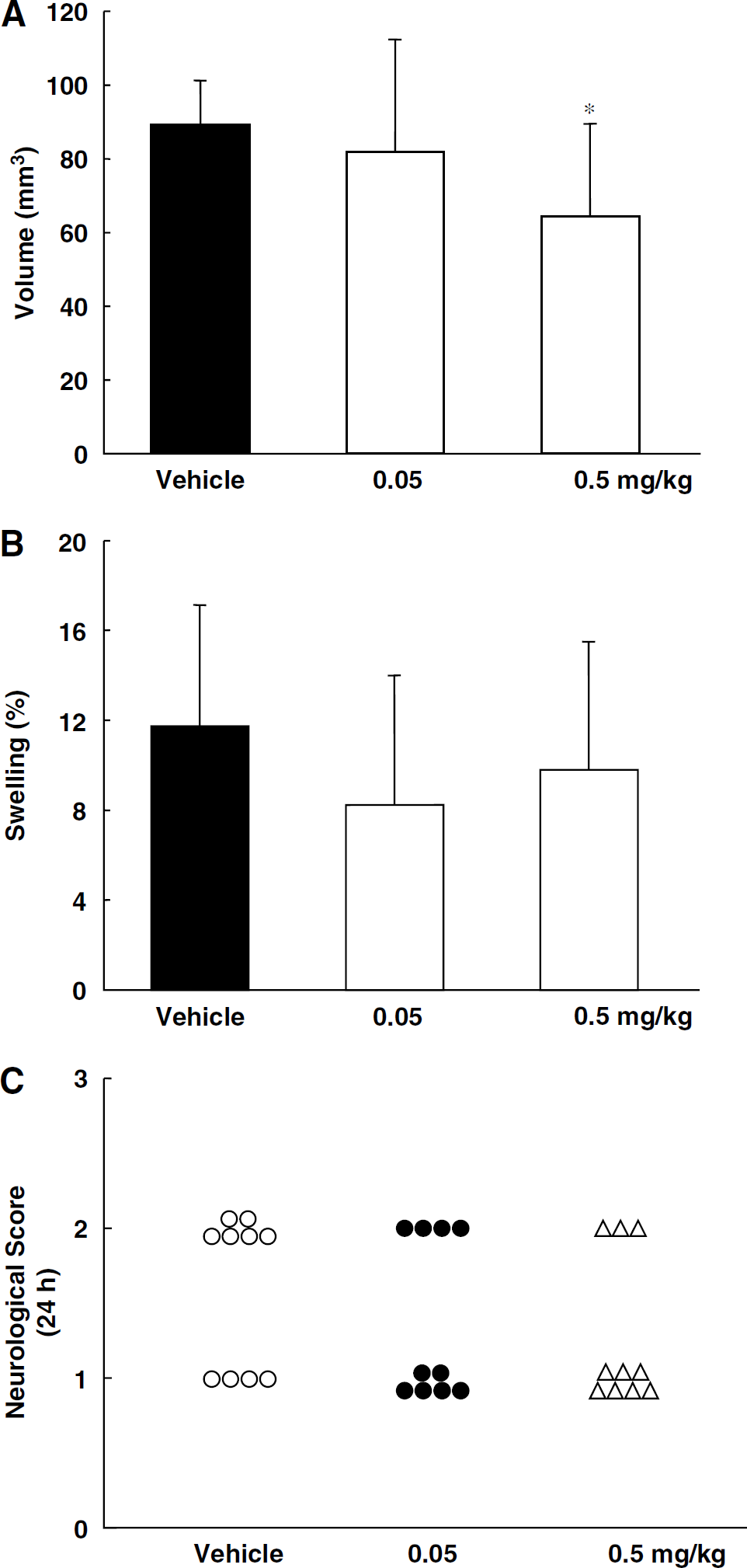
To examine whether EDTA Na might reduce infarct volume, we used TTC staining. Twenty-four hours after MCAO, mice had developed infarcts affecting the cortex and striatum. EDTA Na (0.5 mg/kg intravenously at 10 mins before MCAO) significantly reduced infarct volume, but brain swelling (edema) was not reduced (versus vehicle-treated mice) (Figures 6A and 6B). No significant changes were observed after administration of 0.05 mg/kg EDTA Na intravenously. Administration of 0.05 or 0.5 mg/kg EDTA Na did not reach to a significant level in the scores given for neurologic deficits significantly (Figure 6C).
(
Oxygen—Glucose Deprivation-Induced Apoptosis and Necrosis in PC12 Culture
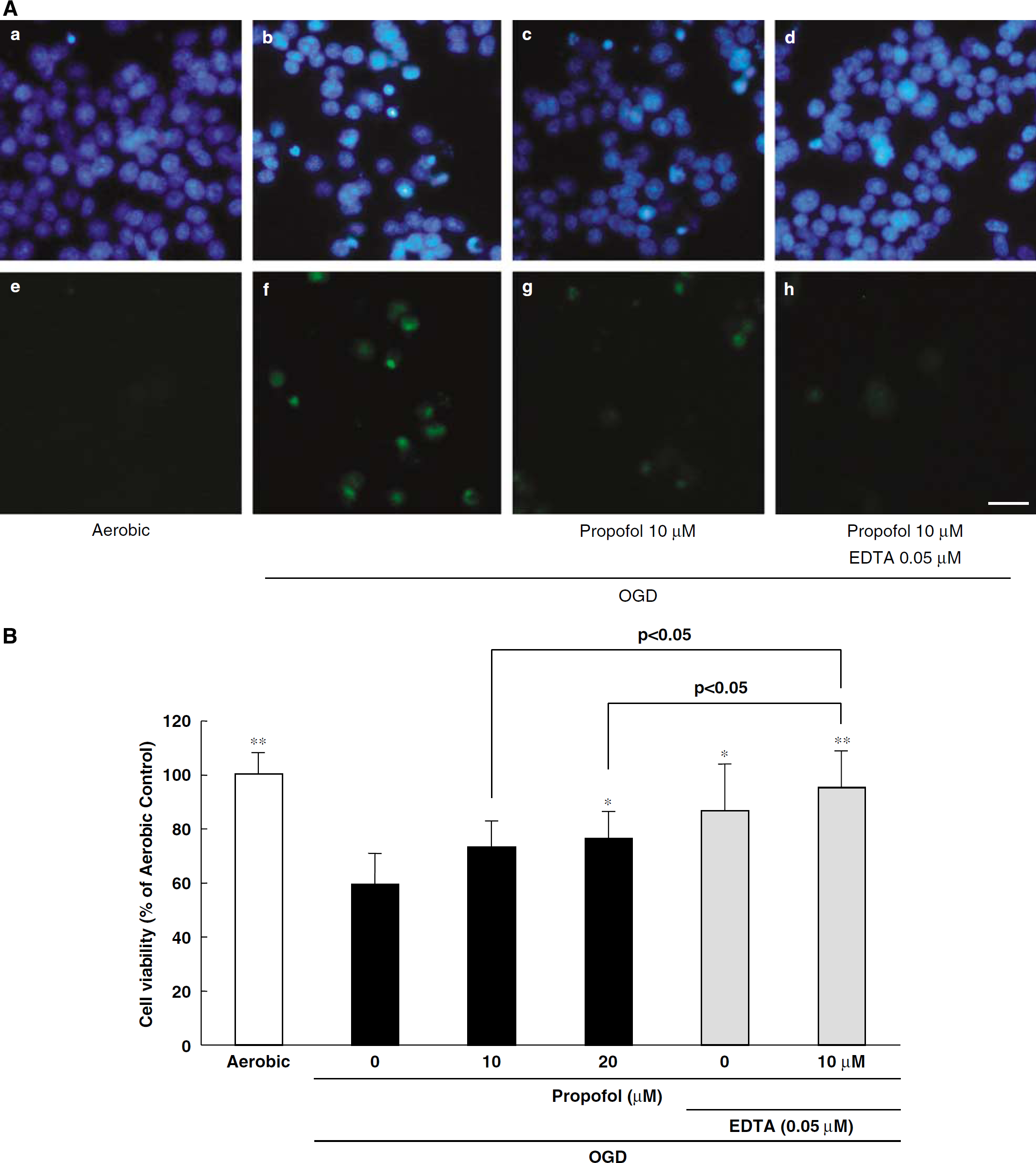
Photographs of Hoechst 33342 and YO-PRO-1 staining are shown in Figure 7Aa to h. Hoechst 33342 stains all cells (live and dead cells), whereas YO-PRO-1 stains early-stage apoptotic and necrotic cells. Propofol or propofol plus EDTA (Figure 7Ag and h, respectively) decreased (versus vehicle treatment) the number of cells showing YO-PRO-1 staining after treatment with OGD. In a resazurin assay for the evaluation of cell viability, cell death was observed at 24 h after OGD treatment. Propofol inhibited OGD-induced cell death in a concentration-dependent manner (as evidenced by its effect on the increase in fluorescence intensity (λex 560/λem 590 nm) associated with the reduction of resazurin to resorufin), its effect being significant at 20 μmol/L (Figure 7B). Similarly, EDTA significantly inhibited cell death at 0.05 μmol/L. Further, propofol plus EDTA inhibited cell death significantly more than propofol alone.
(
Measurement of Intracerebral Zinc Concentrations
Mice were assigned to four experiment groups (see Materials and methods). In the groups that did not undergo MCAO, no changes were found in the intracerebral zinc concentrations in vehicle-treated or propofol EDTA-treated mice. In the propofol EDTA-treated groups that underwent MCAO, the intracerebral zinc concentration in the cortex were decreased significantly (versus vehicle). Conversely, the intracerebral zinc concentration in the subcortical area slightly decreased, but it was not a significant change (versus vehicle). In vehicle-treated mice, there was no significant change in the intracerebral zinc concentrations by ischemia.
Discussion
The main aim of this study was to compare the neuroprotective effects of propofol and propofol EDTA. The neuroprotective effects of propofol are well known, but to our knowledge little is known about neuroprotective effects of EDTA against ischemic damage. Because the chelating action of EDTA was a potential concern, we also examined the effect of propofol EDTA on intracerebral zinc homeostasis during focal cerebral ischemia.
For our in vivo study, we chose a permanent MCAO model because acute ischemic stroke in humans is frequently thromboembolic in nature and rarely undergoes spontaneous reperfusion. Both propofol (10 mg/kg, intravenously) and propofol EDTA (5 to 10 mg/kg, intravenously), when administered 10 mins before the occlusion, led (24 h later) to a smaller infarction, less brain swelling, and improvements in neurologic deficits (versus vehicle) (Figure 1B to 1D). A significant degree of protection was obtained when either drug was administered 10 mins before, but not 1 h after, MCAO (Figures 2A to 2C), with the propofol EDTA group apparently exhibiting the stronger neuroprotective effects. Furthermore, propofol EDTA (10 mg/kg, intravenously), when administered 10 mins before the occlusion, decreased infarct volume and the neurologic deficits significantly 7 days after MCAO (versus vehicle) (Figures 3A and 3B). Adembri et al (2006) reported that propofol reduced the size of the infarct and preserved spontaneous activity, even when its administration was delayed until up to 30 mins after permanentMCAO in rats. In our study, on a different species (mice), only propofol EDTA (not propofol) administration after MCAO reduced the infarct volume significantly, and it did not reduce either the brain swelling or the scores given for neurologic deficits. Moreover, when propofol and propofol EDTA were administered 1 h after MCAO, neither displayed any neuroprotective effects. However, as propofol and propofol EDTA are widely used as anesthetic agents in many surgical procedures and as sedatives for intensive care patients, they will already be present if cerebral ischemia develops during an operation or sedation, and might then provide a valuable neuroprotective effect. However, we have to mention that, in this study, propofol and propofol EDTA protected neurons against ischemia-induced neuronal damage under a hyperglycemic condition (see Table 1). Brown et al (2005) have reported that anesthesia can cause sustained hyperglycemia. The anesthesia or the operation might increase the blood glucose level, presumably by the secreted catecholamine during an operation by ache irritation.
Ischemia-induced TUNEL-positive cells were found in both the ischemic penumbra and core regions in control mice, and propofol EDTA significantly reduced their number in the ischemic penumbra (Figures 5C and 5D). Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling staining detects nuclear DNA fragmentation, which has been reported to occur in both necrosis and apoptosis. Indeed, we observed TUNEL-positive morphologically necrotic neurons mainly in the ischemic core, in addition to apoptotic ones mainly in the inner boundary zone of the infarct (Kitagawa et al, 1998). Thus, our research suggests that propofol EDTA inhibits apoptosis mainly in the ischemic penumbra.
Next, we examined the in vitro effects of propofol and EDTA against OGD-induced cell damage in cultures of PC12 cells (Figures 7A and 7B). Propofol and EDTA each protected PC12 cells against such damage, and the effect of propofol was enhanced when EDTA was added. Thus, both in vitro and in vivo, propofol plus EDTA had a stronger neuroprotective effect than propofol alone. These findings indicate that the protective effects of propofol on ischemic damage are in part due to a neuron-direct mechanism. We therefore examined the neuroprotective effect of EDTA administered alone, and we found that EDTA Na (0.5 mg/kg intravenously) significantly reduced only the infarct volume. Because 0.005% disodium edetate (EDTA) is included in the generally used preparation of propofol EDTA, 10 mg/kg propofol EDTA has 0.05 mg/kg EDTA. A significant neuroprotection was seen with that level of EDTA when 10 times that concentration (0.05 mg/kg) was used. For EDTA to pass from blood into brain tissue, it must cross either the blood—brain barrier or the blood—cerebrospinal fluid barrier; it is well known that EDTA can hardly cross the blood—brain barrier (Fenstermacher and Kaye, 1998). However, the oxidative stress generated during stroke can lead to blood—brain barrier disruption, with secondary vasogenic edema and hemorrhagic transformation of infarcted brain tissue, so that enough EDTA may cross the blood—brain barrier at times of cerebral ischemia to exert neuroprotective effects.
EDTA is a potent chelator of heavy metals such as zinc, iron, copper, manganese, chromium, cobalt, and lead (Herr et al, 2000). We focused on zinc as it is one of the most abundant transition metals in the brain, and is essential for development, growth, DNA synthesis, immunity, and a wide array of cellular processes (Choi and Koh, 1998). The physiologic significance of neuronal zinc release within the central nervous system is not clear, and its role in ischemic brain injury is controversial. After brain ischemia, there is a depletion of presynaptic bouton zinc and a concurrent accumulation of zinc in the cell bodies of vulnerable neurons (Koh et al, 1996). In addition, Sorensen et al (1998) found evidence for early Zn2+ translocation from presynaptic terminals into postsynaptic neuronal cell bodies in the rat cortex subjected to focal ischemia. It has been proposed that synaptic zinc or extracellular zinc acts as ‘the cell-death ion’ during neuronal damage, both in vitro and in vivo (Choi et al, 1998; Shabanzadeh et al, 2004; Koh et al, 1996; Canzoniero et al, 1999). Although some authors believe that zinc may have a protective function (Bancila et al, 2004; Matsushita et al, 1996), it has been shown that when the influx of chelatable extracellular Zn2+ into postsynaptic neurons is blocked by intraventricular injection of a Zn2+-chelating agent, neurodegeneration is prevented (Calderone et al, 2004). However, measurements of the change in intracerebral zinc concentrations are lacking after intravenous administration of EDTA at the time of cerebral ischemia. We therefore administered propofol EDTA (including 0.005% EDTA) intravenously, and measured zinc concentrations with or without cerebral ischemia. When propofol EDTA was administered without ischemia, no change was found in the intracerebral concentrations of zinc. However, the zinc concentration decreased significantly in the cortex, but not in the subcortex, when propofol EDTA was administered 10 mins before MCAO (Table 2). This result indicates that chelation of zinc by EDTA occurs in the cortex during ischemia, possibly resulting in a neuroprotective effect. However, further experiments will be needed to clarify the detailed mechanisms linking zinc to ischemic neuronal damage. Sorensen et al (1998) found that the density of zinc-positive terminals was significantly decreased in the neocortical ischemic zone at 7 mins after MCAO in rats. In addition, Foreman et al (1953) injected CaNa2EDTA into a rat muscle, and found that the metabolic turnover time was approximately 50 mins. On the basis of these findings, we decided to sample brain at 1 h after MCAO. However, we did not try to determine when the level of zinc accumulation in the extracellular space or postsynaptic neuron reaches a peak or when the chelated zinc begins to be egested from the brain.
Zinc levels in mouse brains
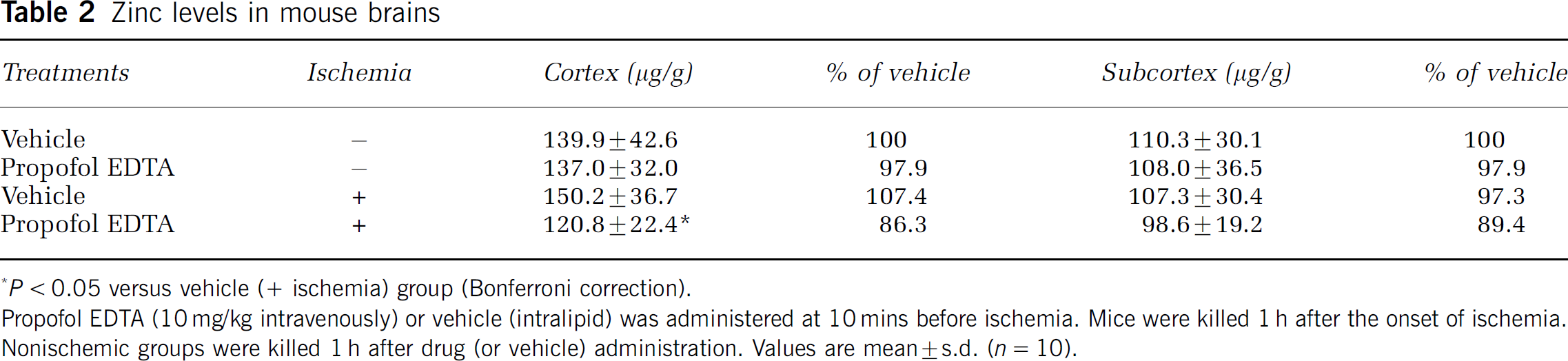
P < 0.05 versus vehicle (+ ischemia) group (Bonferroni correction).
Propofol EDTA (10 mg/kg intravenously) or vehicle (Intralipid) was administered at 10 mins before ischemia. Mice were killed 1 h after the onset of ischemia.
Nonischemic groups were killed 1 h after drug (or vehicle) administration. Values are mean ± s.d. (n = 10).
Second, we did not assess the proportion of zinc chelated by propofol EDTA. Although some investigations have found that Zn2+ chelators protect against ischemic cell death, there is a view that ‘too much extracellular Zn2+ has toxic effects, but some concentrations of Zn2+ have neuroprotective effects’ (Zhao et al, 1996; Kitamura et al, 2006). We therefore need to study the relationship between the proportion of chelated zinc and the associated neuroprotective effects.
Lastly, EDTA can also chelate calcium. An intracellular calcium increase is an early key event triggering ischemic neuronal cell damage (Nikonenko et al, 2005). There is therefore a possibility that EDTA protects cells by modulating calcium influx into the cells, and it is consequently impossible to ascribe the neuroprotective effects of EDTA to a chelation of zinc alone (since we did not measure the concentration of calcium).
In this study, we found that pretreatment with EDTA robustly protects neurons. In principle, chelation therapy using EDTA could be a new treatment for cerebral ischemia. In fact, chelation therapy using intravenous injections of edetate disodium was being promoted to the public two decades ago as a nonsurgical means of treating coronary or other arterial atherosclerosis (Rathmann and Golightly, 1984). However, subsequent researchers have provided no evidence that chelation therapy is efficacious beyond a powerful placebo effect (Ernst, 1997; Shrihari et al, 2006). Moreover, the loss of essential minerals and the possible redistribution of lead within the body may constitute disadvantages that should be taken into account if contemplating repeated intravenous administration of EDTA. Herr et al (2000) compared the safety of propofol with that of propofol EDTA when used for sedation in critically ill postsurgical or trauma patients in the intensive care unit. The propofol EDTA formulation had no effects on calcium or magnesium homeostasis, renal function, or sedation efficacy over and above of propofol alone when used for sedation in critically ill surgical or intensive care unit patients. In addition, Zaloga and Teres (2000) reported that the most notable abnormality in intensive care unit patients given propofol containing EDTA was a low blood zinc level, but no adverse events indicative of zinc deficiency occurred. Such a decrease in the blood zinc concentration by EDTA accords with our result (decrease in the concentration of zinc in brain tissue). However, since EDTA-induced chelation of calcium has the capacity to kill cells, we believe that it is also necessary to consider its concentration when EDTA is administered intravenously.
In summary, propofol and EDTA each inhibited OGD-induced cell damage, cotreatment with propofol and EDTA being more potent than treatment with propofol alone. Propofol EDTA decreased ischemic neuronal damage after MCAO in mice, and its potency was greater than that of propofol alone. These findings indicate that EDTA may itself have a neuroprotective effect, perhaps partially due to its chelation of zinc.