
Abstract
Matrix metalloproteinases (MMPs) disrupt the blood—brain barrier (BBB) during reperfusion. Occludin and claudins are recently described tight junction proteins (TJPs) that form the BBB. We hypothesized that the opening of the BBB was because of the degradation of TJPs by the MMPs. Spontaneously hypertensive rats had a 90 mins middle cerebral artery occlusion with reperfusion for 2, 3, or 24 h. Matrix metalloproteinases were measured by immunohistochemistry and in situ and gel zymography. Real-time polymerase chain reaction (PCR) measured mRNAs of MMP-2 and −9, furin, membrane-type MMP (MT1-MMP), occludin, and claudin-5. There was opening of the BBB in the piriform cortex after 3 h of reperfusion, and an MMP inhibitor, BB-1101 (30 mg/kg), prevented the opening. At 3 h, in situ zymograms showed gelatinase activity. Zymography and PCR showed greater increases in MMP-2 than in MMP-9. There were increased mRNA and immunohistochemistry for MT1-MMP and furin, which activate MMP-2. Claudin-5 and occludin mRNA expression decreased at 2 h in both hemispheres with fragments of both proteins seen on Western blot by 3 h on the ischemic side; treatment with BB-1101 reversed the degradation of the TJPs. Immunohistochemistry at 3 h showed fragmented TJPs within the endothelial cell clefts. By 24 h, in situ zymography showed gelatinase activity and gel zymography showed elevated levels of MMP-9. Disrupted TJPs previously seen in endothelial cells appeared in the surrounding astrocytes. Our results provide direct evidence that MMPs open the BBB by degrading TJPs and that an MMP inhibitor prevents degradation of the TJPs by MMPs.
Keywords
Introduction
Tight junction proteins (TJPs) in endothelial cells are a major structural component of the blood—brain barrier (BBB) formed by components of the neurovascular unit (Ballabh et al, 2004; Bazzoni and Dejana, 2004). During ischemia/hypoxia in endothelial cell cultures and in situ models of BBB, TJPs are disrupted (Huber et al, 2001; Mark and Davis, 2002; Witt et al, 2003; Yamagata et al, 2004). The TJPs include zona occludens (ZOs) proteins in the endothelial cells, while in the endothelial cell clefts are found occludin and claudins (Furuse et al, 1993, 1998; Morita et al, 1999a; Nitta et al, 2003). Proteases of the serine and matrix metalloproteinase (MMP) gene families have been shown to degrade proteins of the basal lamina around the cerebral blood vessels (Hamann et al, 1995; Fukuda et al, 2004). An earlier study showed that gelatinase B (MMP-9) degrades ZOs in focal ischemia (Asahi et al, 2001). Less is known about the effect of MMPs on the major proteins in the endothelial cell clefts, namely, occludin and claudin. Studies of cultures of retinal vessels showed that the blood—retinal barrier is altered by MMP-9 through its action on claudin-5 and occludin (Lohmann et al, 2004; Giebel et al, 2005). Increased expression of MMPs by cultured human umbilical vein endothelial cells induced a shift in the molecular size of occludin from 65- to 50-kDa, which was blocked by the MMP inhibitor 1,10-phenanthroline (Wachtel et al, 1999). An increase in the 50-kDa occludin was seen in cultured human corneal epithelial cells that were treated with active MMP-9 in different concentrations (Pflugfelder et al, 2005). These studies in other tissues suggest that MMPs may affect BBB permeability by acting on the TJPs between endothelial cells as well as on the basal lamina proteins in focal ischemia.
Focal ischemia with reperfusion causes a biphasic opening of the BBB (Kuroiwa et al, 1985). Gelatinase A (MMP-2) has been associated with the initial opening, which takes place within several hours (Chang et al, 2003). This early opening is blocked by broad-spectrum MMP inhibitors, which have been used to block the toxicity of recombinant tissue plasminogen activator (Pfefferkorn and Rosenberg, 2003). Therefore, we hypothesized that disruption of the BBB corresponded with degradation of TJPs, which was mediated by MMPs. We also postulated that MMP inhibitors acted by preventing TJP degradation. To test the hypothesis we studied spontaneously hypertensive rats (SHR) that had undergone a middle cerebral artery occlusion (MCAO) with reperfusion for various times. We measured mRNA for MMP-2 and −9, membrane-type MMP (MT1-MMP), furin, and TJPs. Membrane-type MMP activates MMP-2 and furin activates MT1-MMP (Strongin et al, 1995; Sato et al, 1996). Gelatinases were measured by gelatin and in situ zymography and TJPs by immunohistochemistry and Western blots. Since the interactions between endothelial cells and astrocytes are required for induction of BBB properties, the morphologic relationship of TJPs with astrocytes was analyzed (Duffy et al, 2000; Willis et al, 2004; Garcia et al, 2004).
Materials and methods
Middle Cerebral Artery Occlusion with Reperfusion
The study was approved by the University of New Mexico Animal Care Committee and conformed to the National Institutes of Health Guidelines for use of animals in research. Male SHR, weighing 300 to 320 g, were anesthetized with 1.5% halothane in 70% nitrous oxide and 30% oxygen. At 90-mins MCAO was performed by insertion of an intraluminal nylon suture with a bulb on the end as previously described (Rosenberg et al, 2001). Briefly, neck vessels were exposed through a midline incision, and branches of the right external carotid artery were isolated and ligated. A 6-0 silk suture was loosely tied around the external carotid artery stump. A 4-0-monofilament nylon suture was introduced into the external carotid and advanced into the MCA. A silk suture around the stump was tied down onto the thread with the end of the thread protruding slightly. Reperfusion was achieved by slowly pulling the thread back after 90 mins occlusion. For inhibitor studies, the MMP inhibitor, BB-1101 (Generous gift from A Gearing, British Biotechnology, Oxon, UK), was delivered in 30 mg/kg body weight by intraperitoneal injection 10 mins before the MCAO started. Sham animals underwent neck surgery with a suture inserted, but the suture was rapidly removed.
Blood—Brain Barrier Permeability Observed with Evans Blue and Quantified with 14C-Sucrose
Blood—brain Barrier disruption after reperfusion in MCAO was visualized by injection of 2% Evans blue in saline injected intravenously via femoral vein immediately at the onset of reperfusion in the 3 h reperfusion experiments and 2 h before the end of the 24 h reperfusion experiments. Three or 24 h after reperfusion, the animals were killed, and the brains were removed and rapidly frozen in isopentane at −80°C. Frozen brains were cut into 16 to 20-μm-thick sections with a cryostat and mounted for fluorescent microscopy (Olympus BX51, Olympus Optical Co. Ltd, Japan). Leakage of Evans blue appeared as a red fluorescence.
Brain uptake of sucrose was measured in 10 rats that had undergone 90 mins of MCAO with reperfusion for 3 h. Blood—brain barrier permeability was measured by a modification of the brain uptake method (Rosenberg et al, 1998). At 10 mins before death, the rats were anesthetized with 1.5% halothane in 70% N2O and 30% O2 and infused intravenously over 30 secs with 10 μCi of 14C-sucrose (Dupont/New England Nuclear). Samples of blood were drawn from the femoral vein, and the heart was stopped with an intracardiac injection of a saturated potassium chloride solution. The brains were removed and frozen in 2-methylbutane cooled to −80°C. Tissue sections were taken from the infarcted cortex and caudate and similar noninfarcted regions. Brain and blood samples were dissolved in Aquasol (New England Nuclear) and were counted for radioactivity in a liquid scintillation counter (Packard). The ratio of brain sucrose content to that in the blood was calculated.
Gelatin Zymography
For zymography, tissues were taken from a contiguous 5-mm coronal section directly posterior to the one used for BBB measurements. Similar regions of the infarcted and noninfarcted sides were studied. The frozen tissues were homogenized with lysis buffer (50 mmol/L Tris-HCl pH 7.6, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% Brij-35, 0.02% NaN3, and 1% Triton X-100). The protein concentration in the homogenate was determined using Bradford reagent (Bio-Rad). Matrix metalloproteinase-2 and −9 in homogenates were concentrated with gelatin-sepharose beads (Gelatin Sepharose 4B, Amersham Biosciences, Piscataway, NJ, USA) and analyzed by gelatin zymography as previously reported with modifications (Planas et al, 2000). Briefly, 0.8 mg protein in 500 μl was incubated with 80 μl Gelatin-Sepharose 4B beads for 1 h at 4°C with gentle rotation. The beads were collected and the MMP-2 and −9 were eluted by incubating with 80 μl elution buffer (10% dimethyl sulfoxide in phosphate-buffered saline (PBS) for 30 mins at 4°C with gentle shaking. Equal amounts of samples (20 μl) were electrophoretically separated on 10% sodium dodecyl sulfate -polyacrylamide gels co-polymerized with 1 mg/ml gelatin (Sigma-Aldrich Chemicals, St Louis, MO, USA) under nonreducing conditions. Gels were washed in 2.5% Triton X-100 and then incubated for 40 h with a developing buffer containing 50 mmol/L Tris, pH 7.6, 5 mmol/L CaCl2, 0.2 mmol/L NaCl, and 0.02% (w/v) Brij-35 at 37°C before they were stained with 0.125% Coomassie blue R-250 for 30 mins in 10% (v/v) acetic acid and 50% methanol. Gels were destained with a solution containing 10% acetic acid and 10% methanol until clear bands of gelatinolysis appeared on a dark blue background. To confirm that detected activities were zinc-dependent gelatinases, some zymogram gels were incubated with developing solution in the presence of 10 mmol/L ethylenediaminetetraacetic acid (EDTA). Disappearance of lytic bands in these gels confirmed the metal dependence of gelatinolytic activity characteristic of MMPs. The gels were dried and scanned for densitometry (Alpha Imager™ 2200; Alpha Innotech, San Leandro, CA, USA). A mixture of human MMP-9 and −2 (Chemicon International, Inc. Temecula, CA, USA) was used as gelatinase standards.
In Situ Zymography on Brain Sections with Evans Blue
Sections from brain tissues injected with Evans blue as described above were used for in situ zymography after reperfusion (Rivera et al, 2002). The frozen, nonfixed sections were incubated in a humidity chamber for 3 h at 37°C in a reaction buffer containing 40 μg/ml of fluorescein isothiocyanate (FITC)-labeled DQ-gelatin (EnzCheck Collagenase Kit, Molecular Probes, Eugene, OR, USA). Gelatin-FITC is cleaved by gelatinases, yielding peptides whose fluorescence is representative of net proteolytic activity. Sections were rinsed three times in PBS and mounted in Gel/Mount™ (Biomeda Corp. Foster City, CA, USA) for fluorescence microscopy. In each experiment, some sections were preincubated with blocking antibodies against MMP-2 (dilution 1:300) and MMP-9 (dilution 1:300) for 1 h, or 1 mmol/L 1,10-phenanthroline, a potent zinc chelator that inhibits all MMP activities, in the reaction buffer (Rivera et al, 2002; Frederiks and Mook, 2004). Sections incubated without DQ-gelatin were not fluorescent.
Antibodies Used for Immunohistochemistry and Western Blots
Anti-MMP-2 antibody (Chemicon International, Inc. Temecula, CA, USA) is a polyclonal rabbit anti-rat antibody that recognizes rat MMP-2. Anti-MT1-MMP (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) is a goat polyclonal antibody against a peptide mapping near carboxy terminus of MT1-MMP of human origin. Antifurin antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) is a goat polyclonal antibody against a peptide mapping near carboxy terminus of furin of human origin. Antioccludin and anticlaudin-5 antibodies were purchased from Zymed Laboratories (South San Francisco, CA, USA). Antioccludin (0.25 mg/ml) is a rabbit polyclonal mouse antibody specific for the occludin protein (65 kDa) directed against 150 amino acids in the carboxy terminal region of human occludin. Anti-claudin-5 (0. 5 mg/ml) is a mouse monoclonal antibody that reacts specifically with the 22–24 kDa Claudin-5. Rabbit and mouse monoclonal antiglial fibrillary acidic protein (GFAP) antibodies (Sigma-Aldrich Chemicals, St Louis, MO, USA) were used for localizing GFAP in astrocytes. All of the antibodies above are documented to react with rat by immunoblot and immunohistochemistry. Mouse antineuron-specific nuclear protein (NeuN) (Chemicon International, Inc., Temecula, CA, USA) was used as a marker for identifying neurons. Anti-mouse IgG, anti-rabbit IgG, anti-goat, and anti-rabbit IgG horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laoratories, Inc., USA) were used.
Immunohistochemistry
Immunohistochemistry for MT1-MMP and furin was performed in MCAO rats after reperfusion for 3 h. Ten-μm-thick cryosections of PLP (2% paraformaldehyde, 0.1 mol/L sodium periodate, 0.075 mol/L lysine in 100 mmol/L phosphate buffer at pH 7.3) fixed brains were rinsed in PBS and preincubated in cold acetone for 5 mins. Endogenous peroxidase activity was inhibited with 0.1% H2O2 in methanol for 10 mins. Nonspecific binding sites were blocked by preincubating tissue for 1 h at room temperature in PBS containing 0.1% Tween-20, 1% bovine serum albumin, and 5% normal serum. Sections were then incubated at 4°C overnight in a PBS solution containing the primary antibodies diluted 1:300 (MT1-MMP) or 1:300 (Furin) in PBS, 0.1% Tween-20, 0.5% bovine serum albumin. Tissue was processed as described above with HRP-labeled donkey anti-goat secondary antibodies for 90 mins at room temperature. Immunostaining was visualized with DAB (DAKO Corporation) and sections were counterstained with NeuroTACS™ Blue counterstain (Trevigen Inc., Galthersburg, MD, USA). Double fluorescent staining of MMP-2 (1:300) with MT1-MMP, NeuN, GFAP (1:500), claudin-5 (1 μg/ml), and occludin (1:500) was performed in sections as mentioned above. Sections were rinsed in PBS, preincubated as indicated above without 0.1% H2O2 and then incubated overnight at 4°C with the primary antibodies. Tissue was rinsed and incubated for 2 h at room temperature with secondary antibodies labeled with FITC or Cyanine (CY3), then rinsed and mounted in Prolong Antifade (Molecular Probes, Eugene, OR, USA). Slides were photographed with a fluorescent microscope (Olympus BX51, Olympus Optical Co. Ltd, Japan) and a confocal microscope (Zeiss LSM 510, Carl Zeiss MicroImaging, Thornwood, NY, USA). Sections incubated in the absence of the primary antibody, with rabbit IgGs were not immunofluoresent.
Quantitative Real-Time Polymerase Chain Reaction
Quantitative real-time PCR was performed on total RNA isolated from brain tissues of SHR animals exposed to 90 mins of ischemia with reperfusion for 2 h to quantify the mRNA expression of MMPs, furin, and TJPs. Ten μg RNA was reverse-transcribed with MultiScribe reverse transcriptase and random hexamers (Applied Biosystems, Foster City, CA, USA). Primers for the quantitative realtime PCR are listed in Table 1. Polymerase chain reaction was performed according to the following protocol: 2 mins hold at 50°C, 10 mins hold at 95°C, followed by 15 secs at 95°C, and 1 mins at 60°C (40 cycles). Amplification was quantified using the TaqMan™ probe real-time PCR kit and the SYBR Green kit (Applied Biosystems, Foster City, CA, USA). A linear concentration—response curve was established by diluting pooled samples. Primers and probes for 18S ribosomal RNA (TaqMan Ribosomal RNA Control Reagents, Roche) were also included for quantifying relative gene expression. A GeneAmp 5700 Sequence Detection System (Applied Biosystems) was used to measure the expression of the mRNA levels for MMPs, furin, and TJPs.
Primers for real-time PCR

Western Blots of Tight Junction Proteins
Protein samples of brain tissues extracted by RIPA buffer were analyzed for protein expression of occludin and claudin-5, using Western blot. Samples (150 μg) were separated using a 15% Tris-HCl precast gel for polyacrylamide electrophoresis (Bio-Rad Laboratories, Hercules, CA, USA) at 100 V for 75 to 90 mins. Brilliant Blue R staining solution (Sigma-Aldrich Co., St Louis, MO, USA) was used to normalize protein loading. The proteins were transferred to polyvinylidene fluoride membranes with 100 V at 4°C for 60 mins. The membranes were blocked in PBS containing 5% nonfat milk and 0.5% Tween-20 for 4 h and were then incubated overnight at 4°C with primary antibodies against occludin or claudin-5 at a 1:500 dilution. The membranes were washed with PBS containing 0.5% Tween-20 before incubation with the respective secondary antibodies at a 1:5000 dilution for 75 mins at room temperature. Blots were developed using ChemiLucent Detection System Kit (Chemicon International, Inc. Temecula, CA, USA), and protein bands were visualized on X-ray film. Semiquantitation of the protein was performed with the use of Scion image software (Scion, Frederick, MD, USA), and the results are reported as normalized protein value by using the Brilliant Blue R staining on polyvinylidene fluoride for quantifying relative protein expressions.
Statistical Analysis
For all experiments, data were tested for statistical significance with a two-tailed unpaired t-test or a one-way ANOVA for multiple t-tests (Prism, GraphPad Software Incorporated, San Diego, CA, USA). The statistical analysis was run on the normalized data for real-time PCR and Western blot to determine significant differences. The statistical significance was set at P < 0.05. The data are expressed as mean ± standard error of the mean (s.e.m.).
Results
Matrix metalloproteinase-2 Gelatinolytic Activity in Piriform Cortex Corresponds with Early Opening of Blood—Brain Barrier
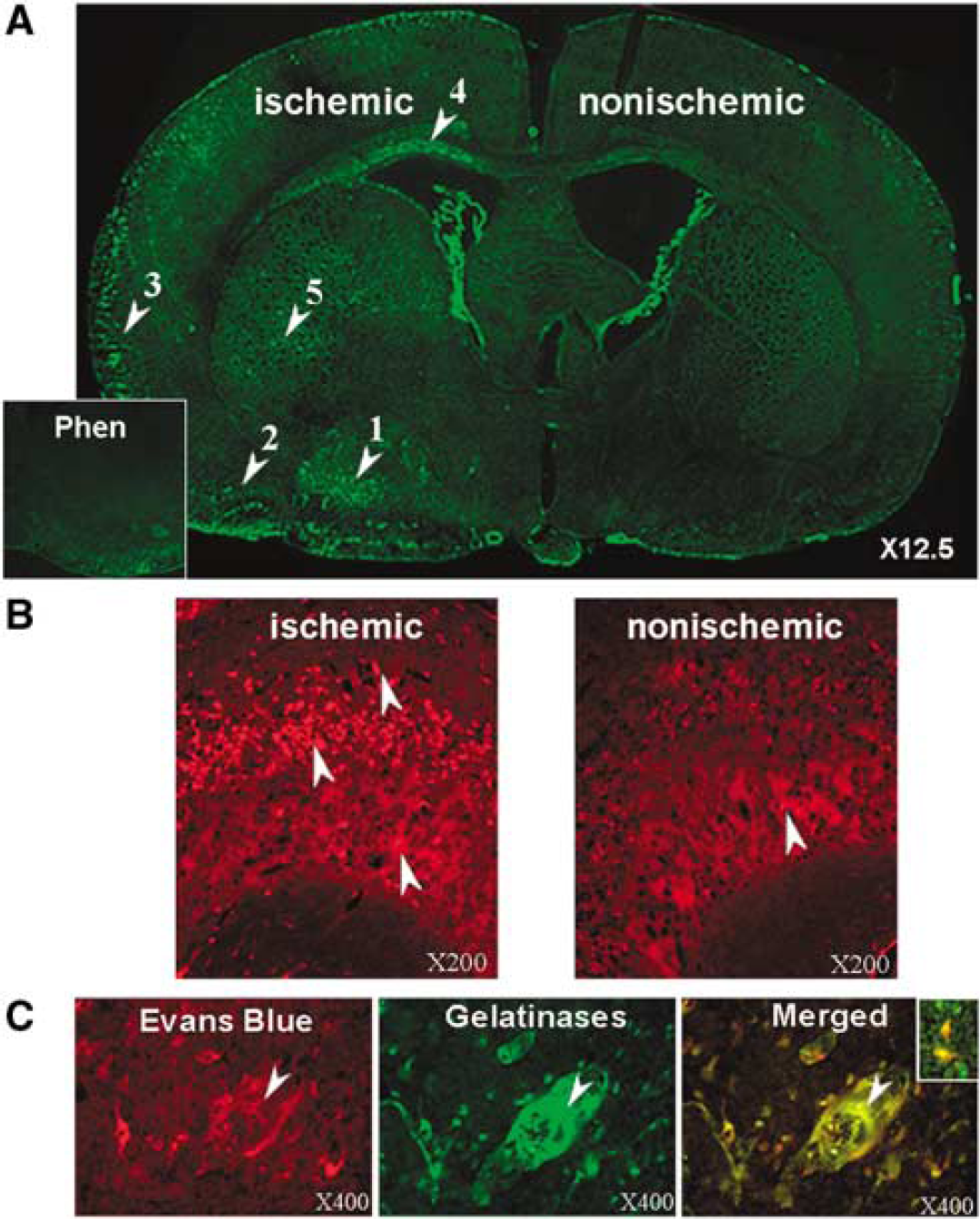
We used in situ zymography to identify the regions with gelatinolytic activity. After 3 h of reperfusion, there was a marked increase in gelatinase activity in the ischemic hemisphere that was mainly seen in the piriform cortex, corpus callosum, caudate, and lateral cortex near the meninges (Figure 1A). Preincubation of MMP-2 antibody reduced the fluorescence signal, and the metalloproteinase inhibitor, 1,10-phenanthroline, also reduced postischemic gelatinase activity (Figure 1A, inset).
(
Matrix metalloproteinase-2 immunostaining showed increased expression in the piriform cortex in the ischemic, but only minimally on the nonischemic side (Figure 1B). The Evans blue was seen grossly in the region of the piriform cortex where the gelatinase activity was prominent. To further confirm that the leaky vessels were associated with the increased gelatinolytic activity, we co-localized the fluorescence from the Evans blue and the in situ zymography. Sites of increased Evans blue signal in the ischemic hemisphere co-localized with the regions of gelatinase activity (Figure 1C).
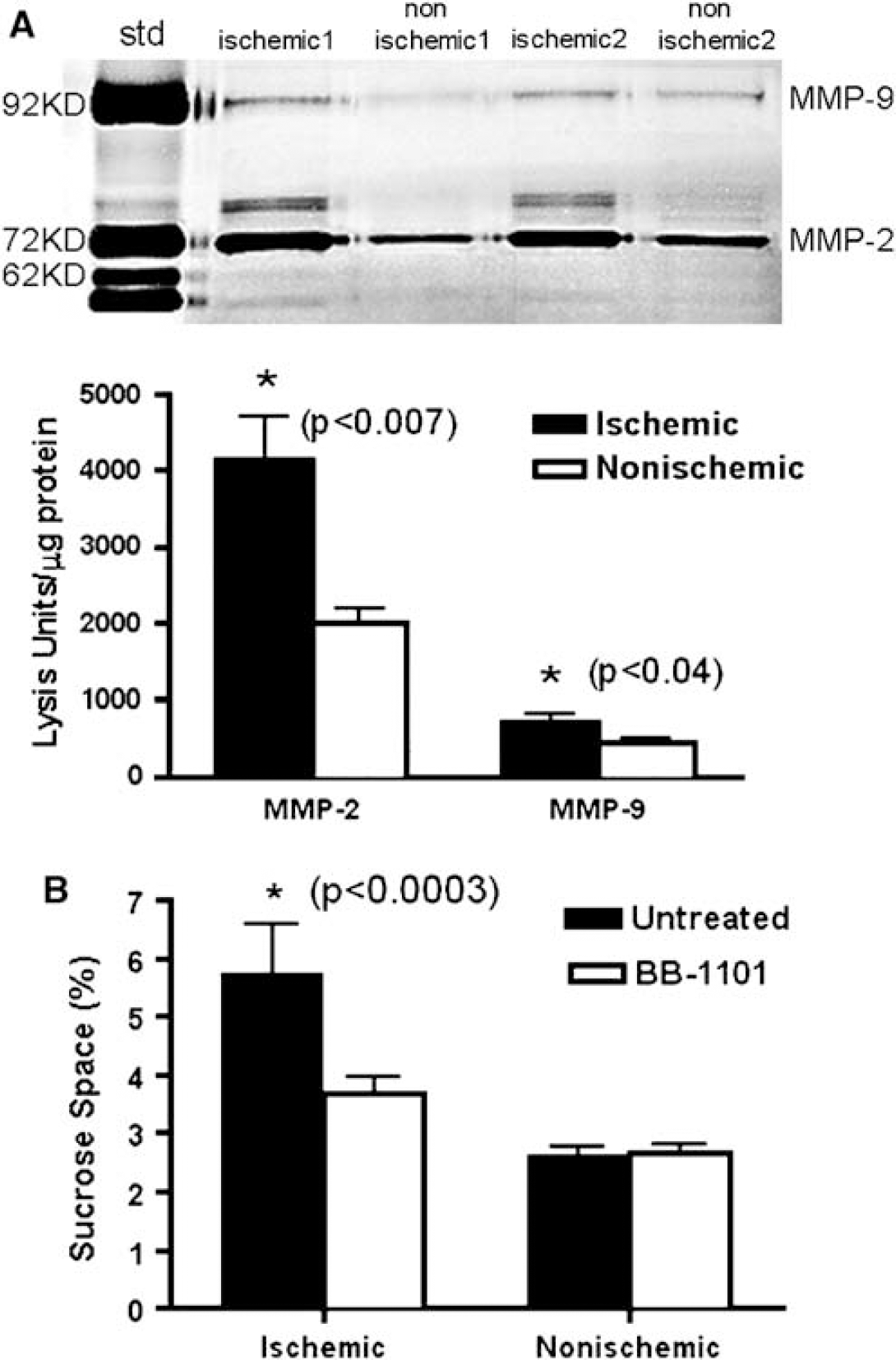
As in situ zymography cannot distinguish between MMP-2 and −9, gelatin-substrate zymography was used to identify the source of gelatinase activity. Tissue samples from the piriform cortex showed a significant increase in the 72-kDa MMP-2 (P < 0.007), and a smaller, but significant increase in 92-kDa MMP-9 (P < 0.04) compared with the nonischemic side (Figure 2A). A 62-kDa form of the MMP-2 proform was seen in the ischemic tissues, indicating activation of MMP-2. There were no active bands of MMP-9 observed in the zymograms.
(
Matrix Metalloproteinase Inhibitor Reduced the Blood—Brain Barrier Opening
Qualitative evidence for disruption of the BBB particularly in the piriform cortex was obtained with Evans blue. We quantified the BBB injury in the piriform cortex, using the brain uptake of 14C-sucrose. After intravenous injection of the radiolabel, tissue samples were collected from the piriform cortex. We found a marked increase of the sucrose space in the ischemic hemisphere. The broad-spectrum MMP inhibitor, BB-1101, significantly reduced the BBB opening on the ischemic side (P < 0.0003) without affecting the nonischemic hemisphere, implicating MMPs in the increased vascular permeability (Figure 2B).
mRNA of Matrix Metalloproteinase-2 Activators Increased at 2 h
Matrix metalloproteinase-2 is secreted in a latent form that requires activation at the cell surface by the action of MT1-MMP, which is activated by the proconvertase, furin. We used real-time quantitative PCR to measure mRNA for MMP-2, MT1-MMP, and furin. By 2 h of reperfusion, there was an increase in mRNA for MMP-2 (Figure 3A). In addition, we found an increase in mRNA for MT1-MMP (Figure 3C), and furin (Figure 3D) in the piriform region of the ischemic hemisphere. At this early time point, there was no change seen in the mRNA for MMP-9 (Figure 3B).
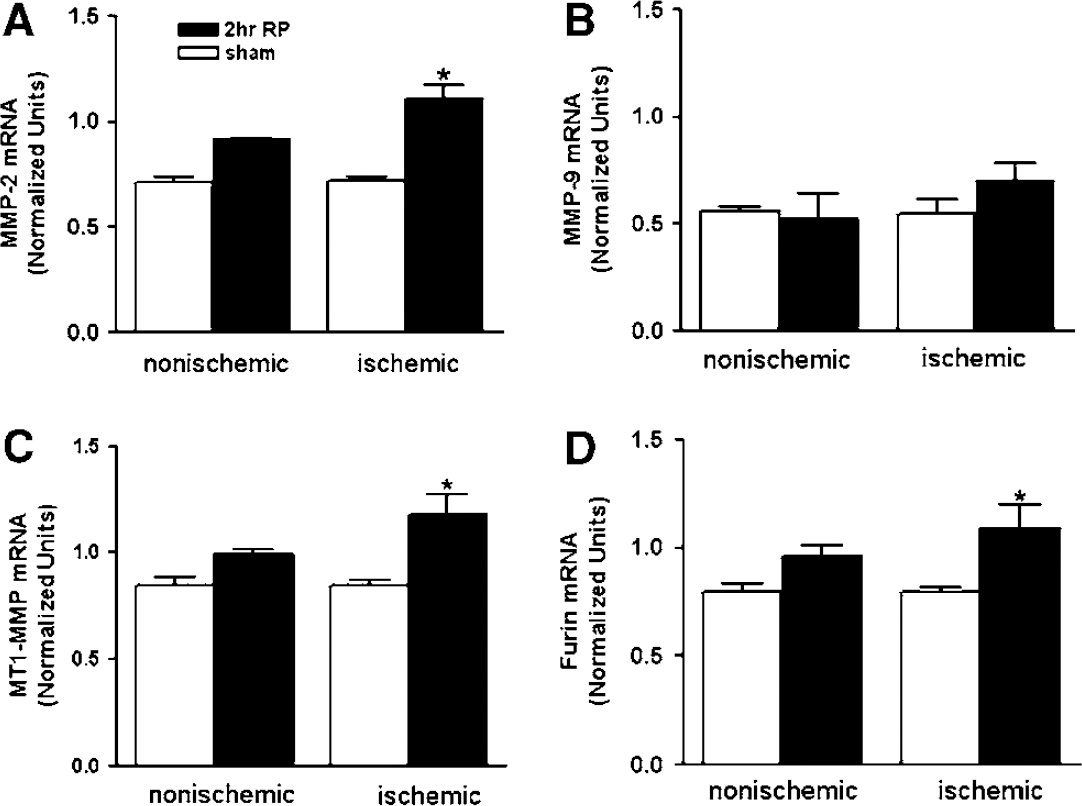
Quantitative real-time PCR for mRNA expression of MMP-2, −9, MT1-MMP, and furin in animals with a 90-min MCAO and 2 h reperfusion (2 h RP). (
Immunohistochemistry Showed Matrix Metalloproteinase-2 in Astrocytes and Neurons
Immunohistochemistry for MMP-2 was performed to determine the cell types expressing the protein. There were very low levels of MMP-2 expression in nonischemic hemisphere, but increased MMP-2 immunoreactivity was seen in the ischemic hemisphere. To determine the cellular sources involved in increased expression of MMP-2, sequential double fluorescent immunostaining was observed by confocal microscopy. A strong association of MMP-2 immunoreactivity with GFAP staining was observed in the piriform cortex, corpus callosum, caudate, and the lateral cortex near the meninges, suggesting that astrocytes are an important source of MMP-2 (Figure 4A). Matrix metalloproteinase-2 co-localized with GFAP-expressing astrocytes in the region of the end feet next to the piriform cortex blood vessels where gelatinolytic activity was seen with in situ zymography and Evans blue leakage was also prominently observed. NeuN staining showed that gelatinase activity was located in neurons in lateral and piriform cortices (Figure 4B). MT1-MMP immunostaining co-localized with MMP-2 in astrocytes, where it was seen mainly around the periphery of the cells (Figure 4C).
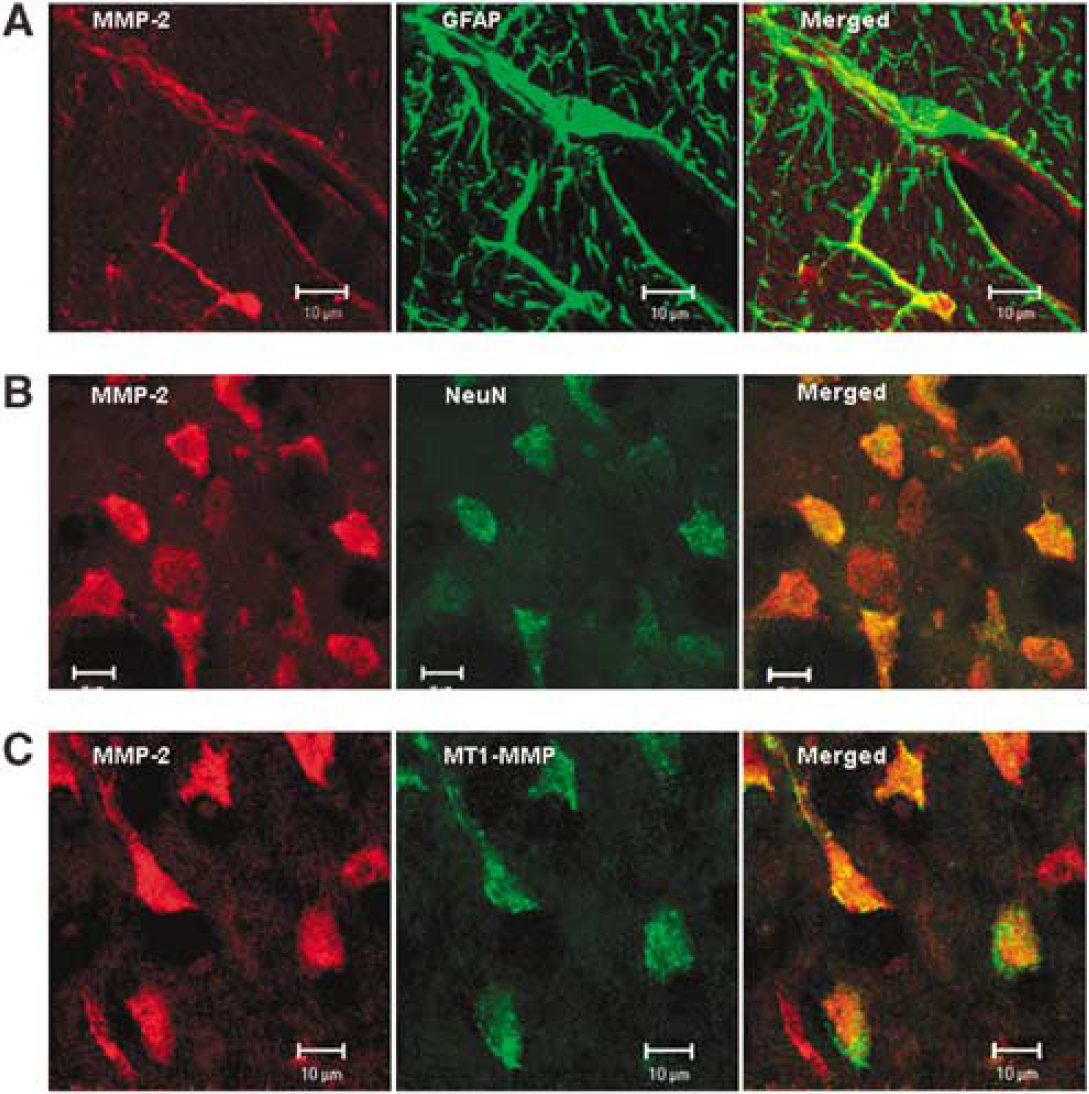
Confocal micrographs showing the immunostaining for MMP-2 and MT1-MMP in glial cells and neurons in the ischemic hemisphere. (
DAB immunostaining showed the MT1-MMP around the blood vessels in the ischemic side in regions similar to those that showed staining for furin, while both were absent on the nonischemic side (data not shown).
Reorganization of Claudin-5 and Occludin Occurs Rapidly in Reperfusion Injury
The effect of focal ischemia on TJPs was studied by immunohistochemistry, PCR, and Western blot. We studied the two major proteins involved in the tight junctions of cerebral vascular endothelia, claudin-5 and occludin. Claudin-5 was found in blood vessels in the sham-operated animals, forming straight rows of staining (Figure 5A). Astrocyte end feet around the blood vessels were completely separated from the claudin-5. Claudin-5 immunoreactivity in nonischemic cortex was similar to that seen in sham-operated animals (Figure 5B). Focal ischemia caused a fragmentation of the claudin-5 (Figure 5C). This resulted in a decrease in the intensity of the claudin-5 staining on the ischemic side, which was absent in some vessels, suggesting that the claudin-5 was reorganized or broken down.
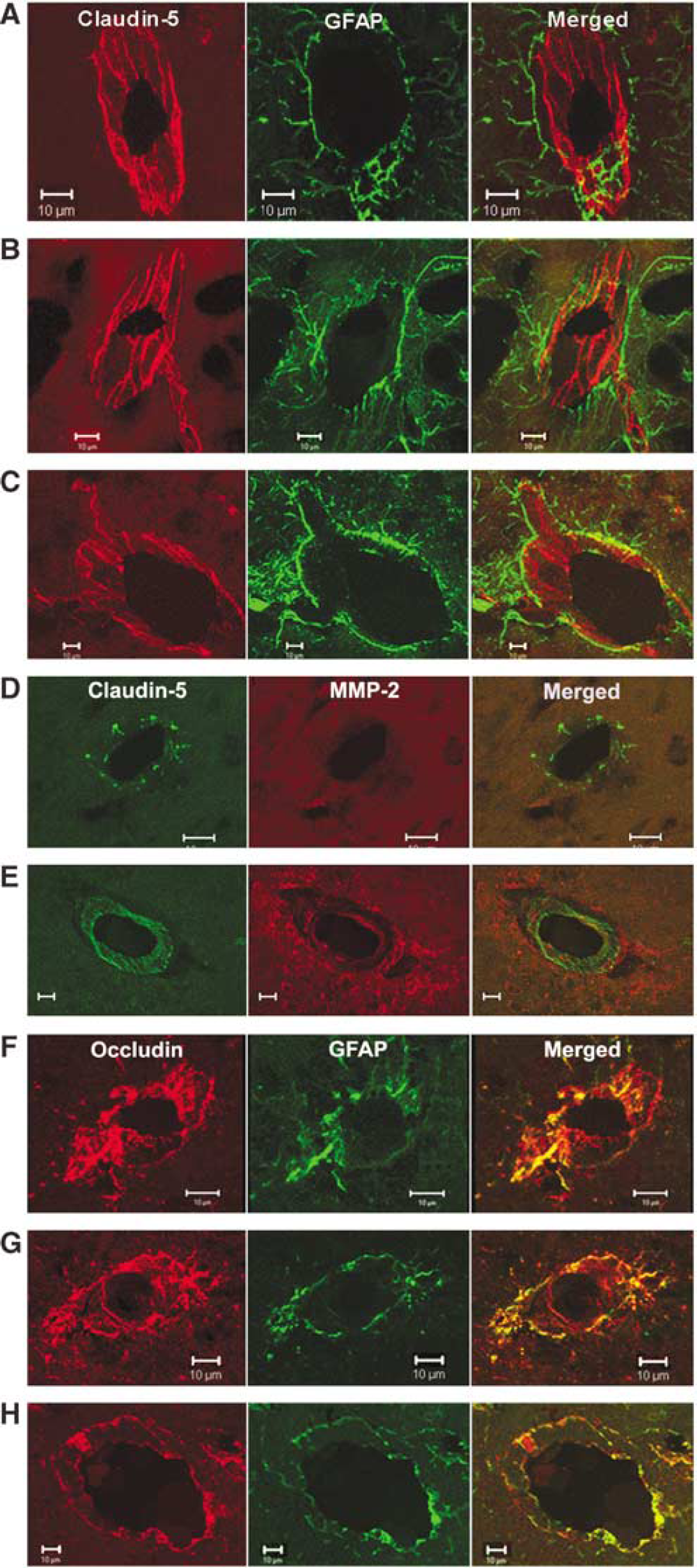
Confocal micrographs showing claudin-5 and occludin immunoreactivity in the sham-operated animals and nonischemic hemispheres after 3 h of reperfusion. (
To further confirm that the fragmentation of the claudin-5 was associated with the increased MMP-2 activity, we co-localized the fluorescence from MMP-2 and claudin-5. In the nonischemic hemisphere, a cross-section of a blood vessel immunostained for claudin-5 shows the rod-like linear strands of protein as dots (Figure 5D). Matrix metalloproteinase-2 is absent from the region of a blood vessel in the nonischemic hemisphere, which is seen more clearly in the merged image (Figure 5D). In the ischemic hemisphere, a blood vessel sectioned at an angle shows attenuated strands of claudin-5 and increased MMP-2 immunoreativity surrounding a vessel. The merged image shows the co-localization of the MMP-2 around the claudin-5 (Figure 5E).
Occludin, another integral TJP, anchors the claudins to ZO-1 and ZO-2, and the plasma membranes of adjacent cells. We observed a continuous distribution of occludin immunostaining in the cerebral vessels in the sham-operated animals, which co-localized with GFAP-positive astrocytes (Figure 5F). A similar pattern of occludin immunostaining was seen in nonischemic cortex vessels (Figure 5G). However, in the ischemic piriform cortex, cerebral vessels showed a reduction in the intensity of occludin immunostaining, suggesting degradation of occludin protein (Figure 5H).
Real-time quantitative mRNA analysis at 2 h reperfusion for claudin-5 (Figure 6A) and occludin (Figure 6B) showed a statistically significant drop in the levels of both TJPs in the ischemic and nonischemic hemispheres.
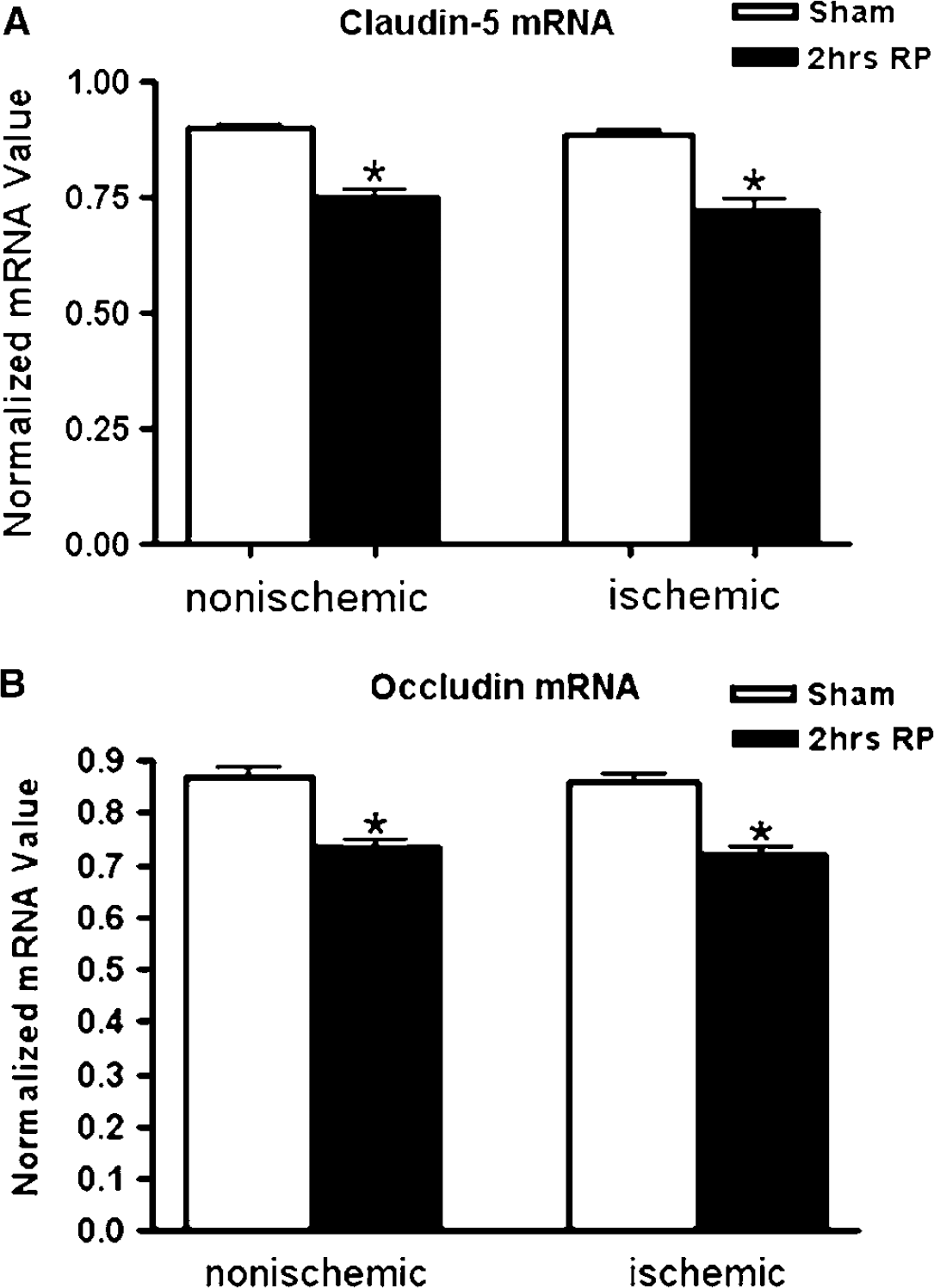
Real-time PCR for the expression of TJP mRNAs. (
Loss of Claudin-5 and Occludin Protein Reversed by A Matrix Metalloproteinase Inhibitor
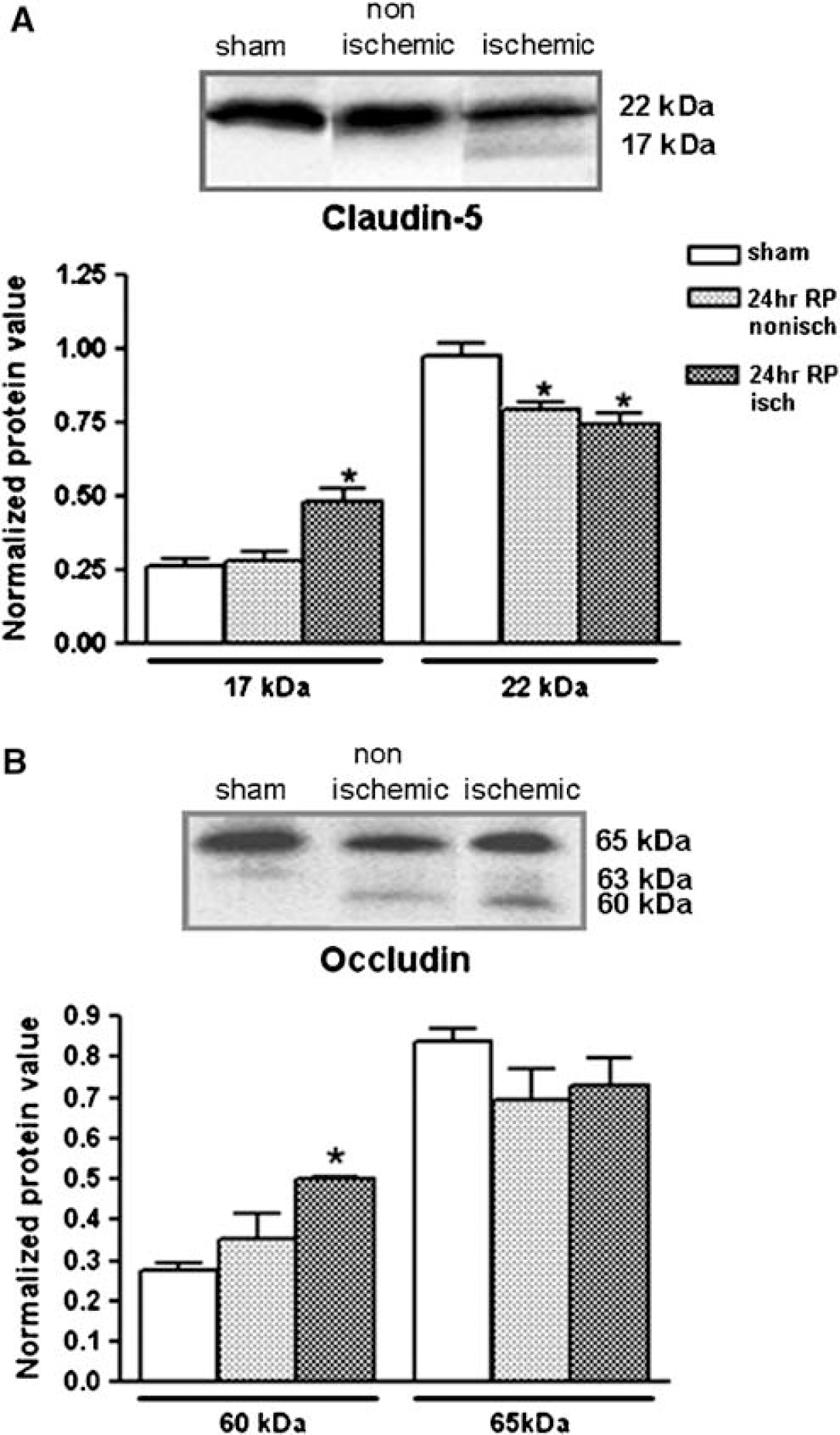
Claudin-5 has a molecular weight normally of 22-kDa. When it is degraded, there are lower molecular weight fragments seen. Immunoblots of TJPs after 3 h of reperfusion showed a marked drop in 22-kDa claudin-5 (P < 0.01) in the ischemic hemispheres (Figure 7A). In addition, reperfusion resulted in a significant increase in the 17-kDa claudin-5 fragment in the ischemic hemisphere (P < 0.01) (Figure 7A), suggesting that the claudin-5 molecule underwent degradation as observed in the claudin-5 immunostaining.
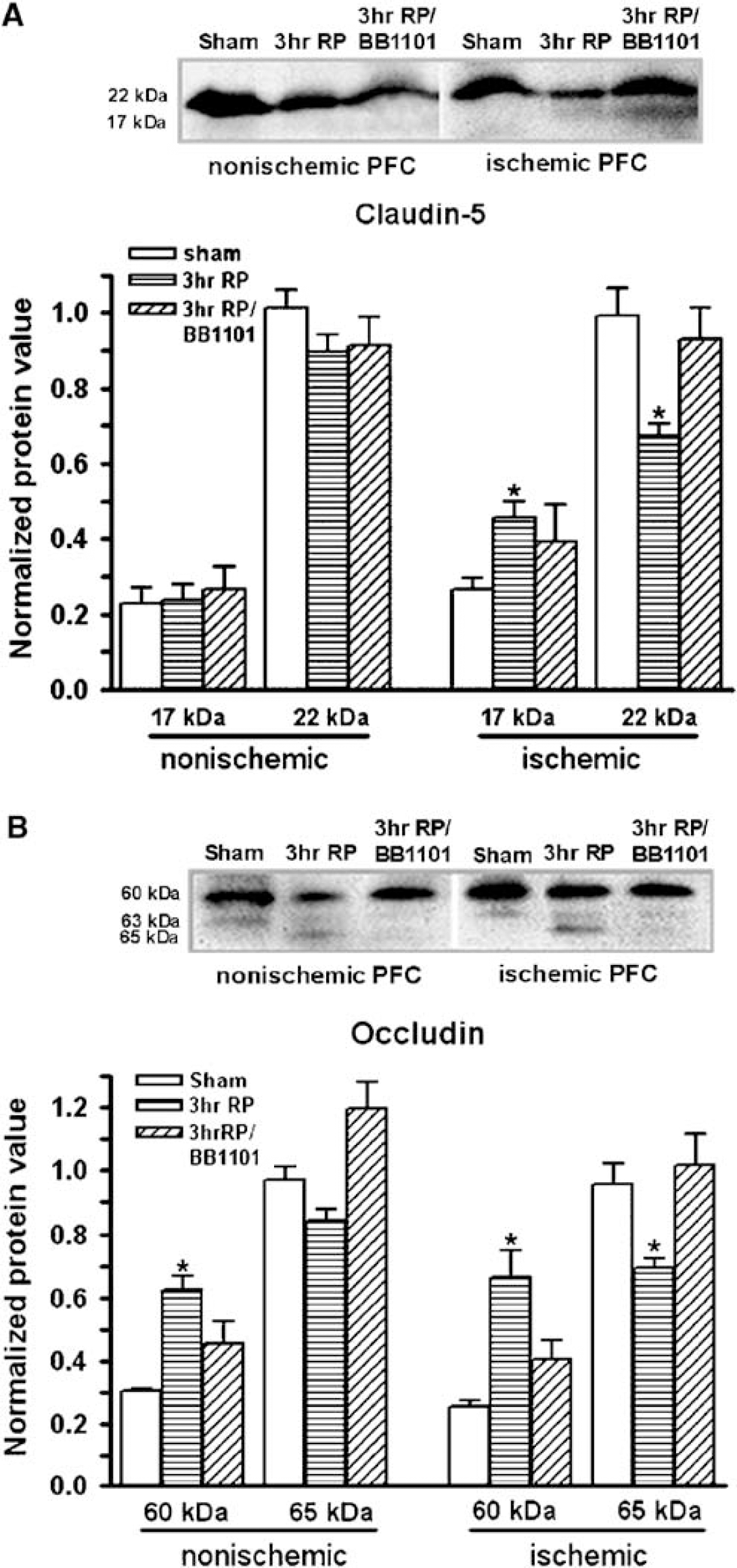
Western blots showing degradation of TJPs after a 90-min MCAO followed by 3h reperfusion (3 h RP). (
Changes in expression of occludin after 90 mins MCAO with 3 h reperfusion were also demonstrated by Western blot (Figure 7B). Occludin normally has bands at 65- and 63-kDa (Figure 7B), which is indicative of phosphorylated (β-band) and nonphosphorylated (α-band) states, respectively (Witt et al, 2003). A significant reduction in the 65-kDa occludin band was seen by Western blot in the ischemic side (P < 0.002). A concomitant increase in the occludin fragment at a molecular weight of 60-kDa was seen in the blots in both nonischemic and ipsilateral hemispheres at 3h reperfusion (P < 0.003 and 0.02, respectively).
Since BB-1101 blocked the early opening of the BBB, we tested the effect of this broad-spectrum inhibitor on prevention of degradation of TJPs with Western blots. Quantification by densitometry showed that the reversal in the fall in the higher molecular weight band of claudin-5 was statistically significant, providing strong support for a major role of the MMPs in TJP reorganization (Figure 7A). Furthermore, BB-1101 significantly blocked the reduction in expression of occludin at 3 h reperfusion and the appearance of the third band (Figure 7B), indicating that MMPs induced occludin proteolysis after MCAO with reperfusion, but had a more complicated effect on claudin-5.
Matrix Metalloproteinase-9 Protein and mRNA Associated with Blood—Brain Barrier Opening at 24 h
At 24 h mRNA for MMP-9 increased without any changes in the MMP-2 mRNA, which was the reverse of the pattern found at 3 h (Figure 8A and 8B). Gelatin gel zymography confirmed that MMP-9 was significantly increased on the ischemic side, while the MMP-2 was not significantly altered (Figure 8C). The increased MMP-9 was associated with disruption of the BBB on the ischemic side (Figure 8D).
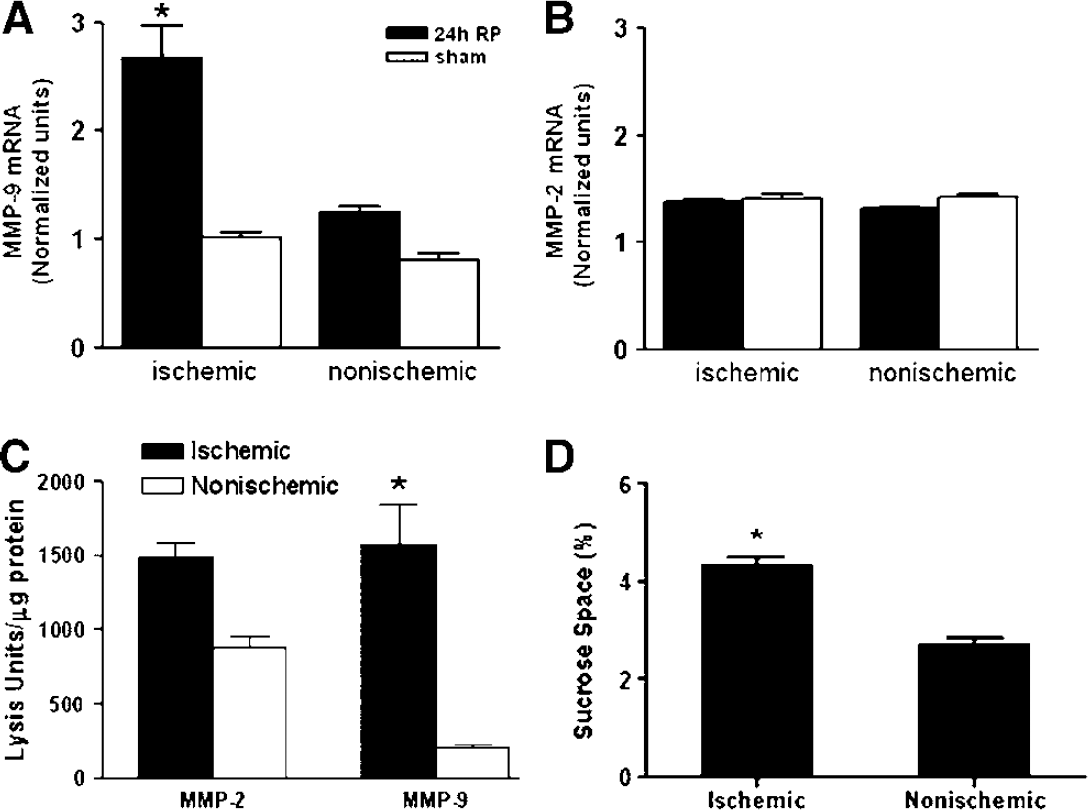
(
In the animals studied after 24 h of reperfusion, we observed intense gelatinase activity in the in situ zymograms in the gray matter of the lateral and piriform cortices and lateral caudate (Figure 9A). Preincubation of the tissue sections with antibodies against MMP-2 and −9 dramatically reduced the fluorescence, showing that the gelatinolytic activity was from either MMP-2 or −9 (Figure 9A, insets).
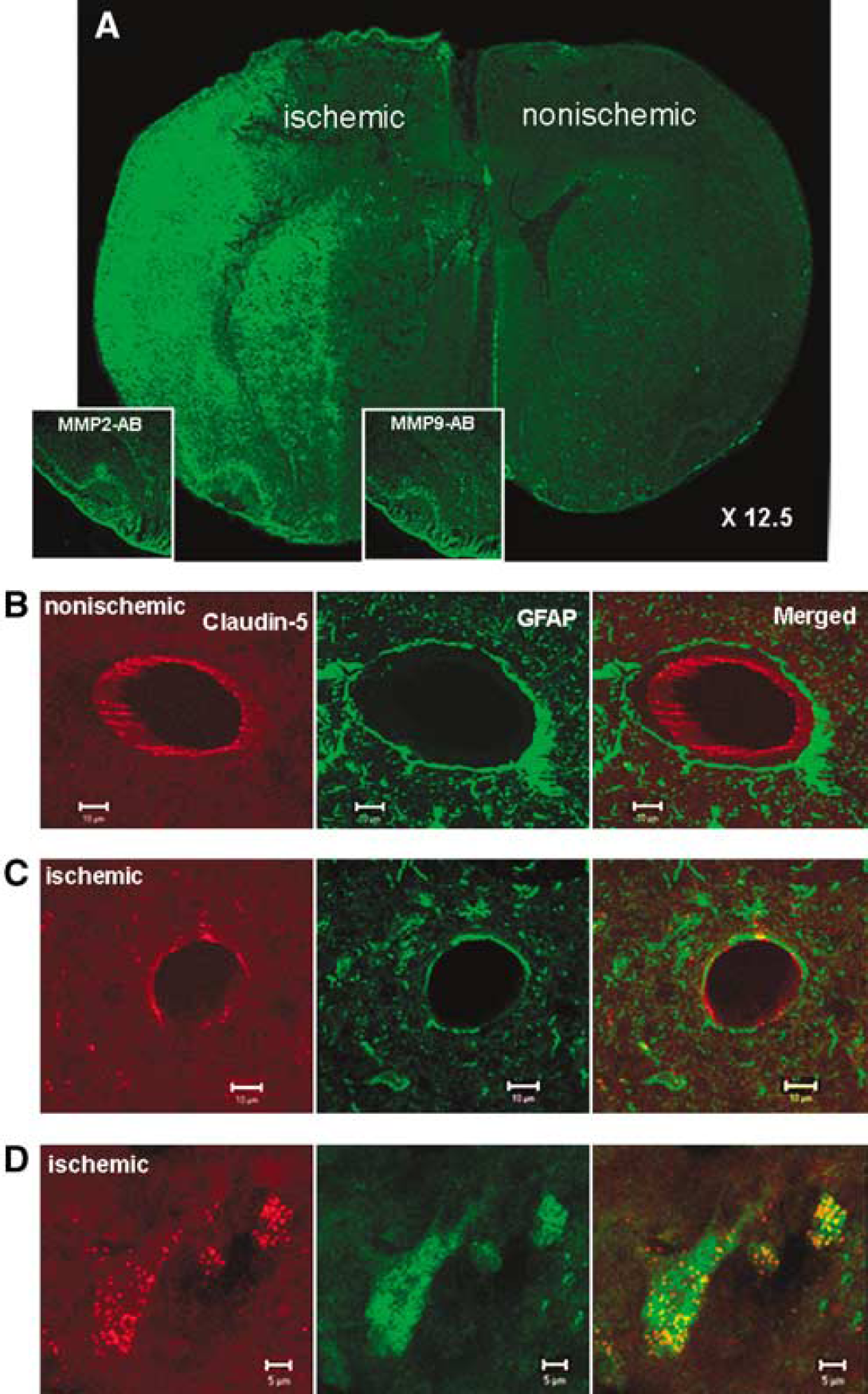
(
Claudin-5 and Occludin Translocate to Astrocytes at 24 h
A different pattern of claudin-5 and occludin immunoreactivity was seen at 24 h in the ischemic hemisphere. Blood vessels on the nonischemic hemisphere showed the characteristic linear patterns of claudin-5 with clearly marked separations between the GFAP-positive astrocytic foot processes and the claudin-5 immunostaining and a continuous rim or line of immunostaining in the endothelial cells (Figure 9B). However, ischemia caused a loss of the claudin-5 immunoreactivity of blood vessels, and a disrupted appearance of astrocytic foot processes around the endothelial cells (Figure 9C). We observed GFAP-positive immunoreactivity co-localized with a diffuse cytoplasmic claudin-5 immunostaining on the ischemic hemisphere in the lateral and piriform cortices (Figure 9D). Similar results were obtained when sections were double labeled for occludin and GFAP (data not shown).
Western blots showed a significant reduction of 22-kDa claudin-5 in both hemispheres at 24 h (P < 0.05, 0.01, respectively, n = 6; Figure 10A). There was a significant increase in the 17-kDa fragment of claudin-5 that was only present in the ischemic hemisphere (P < 0.002). On the contrary, the decrease in occludin, although present in both hemispheres failed to reach statistical significance, but there was a significant increase in the lower molecular weight fragment (P < 0.0001; Figure 10B).
(
Discussion
The early opening in the biphasic pattern of BBB changes seen after reperfusion has been shown to be important in the toxicity of recombinant tissue plasminogen activator, and closing the BBB with inhibitors to MMPs dramatically reduces the toxicity. Our results show that there is a structural change in the proteins forming the tight junctions of the BBB that leads to increased permeability and that a broad-spectrum MMP inhibitor prevents the changes in the TJPs. We found that the early change in permeability was related to an increase in mRNA and protein of MMP-2, and that the major activator of MMP-2, MT1-MMP, and the activator of MT1-MMP, furin, were also increased. More importantly, there was a change in the gelatinolytic activity as measured by in situ zymography in the regions where the TJPs were disrupted. Western blots showed that the TJPs were degraded both at the early time point and in the delayed injury to the BBB, and that the degradation was partially reversed by the MMP inhibitor. The TJPs remained within the endothelial cell clefts during the initial opening and appeared to be attenuated or fragmented. However, a strikingly different pattern of TJPs was seen in the brains of the animals reperfused for 24 h. We observed that the TJPs, which were decreased in the endothelial cells, were co-localized with astrocytes in the region of the vessels. This could explain the reversibility of the early opening and the more prolonged injury associated with the delayed injury to the BBB. Our results suggest that the initial reversible opening of the BBB is related to activation of the constitutively expressed MMP-2, and that the TJPs may be loosened, but remain within the endothelial clefts. However, with lapse of time and increased neuroinflammation resulting in the expression of MMP-9 and possibly other proteases and free radicals, the TJPs are further degraded in the endothelial cell clefts. These results provide a molecular basis for both the transient and long-term alterations in the BBB due to the MMPs in reperfusion injury, and they provide a molecular explanation for the beneficial effects of MMP inhibitors at the early reperfusion times.
Earlier studies provided indirect evidence that MMP-2 played a key role in the initial opening of the BBB in rat brain (Rosenberg et al, 1998), and in the non-human primate brain (Heo et al, 1999; Chang et al, 2003). We combined in situ zymography, which identified the regions where the gelatinases, either MMP-2 or −9, were active, and gel zymography, which showed that the major form was MMP-2. In addition, the presence of mRNA and protein for the activators of MMP-2, namely, MT1-MMP and furin, further supports that MMP-2 was the major enzyme involved in the early TJP disruption. These findings support the studies in the non-human primate (Fukuda et al, 2004). Immunohistochemistry showed that MMP-2 protein was markedly expressed in the ischemic regions in both neurons and astrocytes. We co-localized the MMP-2 and MT1-MMP in the neurons and the astrocytic foot processes. While the MMP-2 in the astrocytic foot processes abutting the cerebral blood vessels most likely contributes to the changes in BBB permeability, the role of the MMP-2 in the neurons remains to be elucidated.
A very different picture emerged at 24 h where the pattern of gelatinase activity in the in situ zymography was much more extensive and the dominant gelatinase expressed in gelatin zymograms was MMP-9. There was expression of MMP-9 mRNA, but not that of MMP-2, which was consistent with the protein expression. Earlier studies had demonstrated expression of MMP-9 at 24 h (Rosenberg et al, 1996; Romanic et al, 1998; Gasche et al, 1999). A striking feature of the later injury to the BBB was the appearance of the TJPs in the astrocytes. Further studies will have to be performed to determine the source of the TJPs in astrocytes.
Tight junction proteins join endothelial cells together to form the interface between blood and brain (Ballabh et al, 2004). In brain, claudin-1 and claudin-5, together with occludin have been described to be present in endothelial TJs forming the BBB and to be both required for formation of the BBB (Morita et al, 1999b; Liebner et al, 2000). Claudins form the primary seal of the tight junction. Claudin-5 is a 22-kDa phosphoprotein with four transmembrane domains. Loss of claudin-1 from cerebral vessels was demonstrated under pathologic conditions such as tumor, stroke, and inflammation (Liebner et al, 2000; Lippoldt et al, 2000). Occludin is a 65-kDa phosphoprotein that assembles with claudin-5 into heteropolymers and intramembranous strands; it is a regulatory protein that can alter paracellular permeability (Tsukita and Furuse, 2000; Hirase et al, 2001; Hawkins and Davis, 2005). Zona occludens protein was shown to be degraded in ischemic injury in the mouse brain, but occludin was found to be unaffected (Asahi et al, 2001). Breakdown of BBB in tissue surrounding brain tumors occurs with concomitant loss of occludin expression (Papadopoulos et al, 2004). In vitro, hypoxia with reoxygenation altered occludin location and molecular weight, which correlated with the observed changes in cerebral microvascular permeability (Mark and Davis, 2002; Witt et al, 2003).
Cerebral blood vessels in the nonischemic piriform cortex were immunostained with claudin-5, which has been described in cerebral endothelial cells. The claudin-5 appeared in linear rows, which has been referred previously to have a ‘zip-locked' appearance (Tsukita et al, 2001). Several possible mechanisms could explain the apparent loosening of the tight junctions. MMPs are released by astrocytic foot processes where they could degrade the basal lamina proteins, which may be providing structural support for the capillary. Another possible explanation is that the attack on the basal lamina exposes the TJPs, which are hydrolyzed by MMPs contained in the blood vessels, such as, MMP-9. The fall in TJP mRNAs in both hemispheres suggests a generalized suppression of synthesis because of the ischemic insult. Another possibility is a decrease in stability of the mRNAs. Further studies will be needed to clarify this issue.
The second opening of the BBB at 24 h is more complicated and results in greater tissue damage. By that time the inflammatory response secondary to ischemia/hypoxia with reperfusion has fully developed, and many additional factors are involved, including cytokines, MMP-3 and −9, and infiltrating leukocytes (Lo et al, 2003). Matrix metalloproteinase-9 is released by several types of cells as well as by infiltrating neutrophils and lymphocytes, making it a highly toxic substance involved in the induction of edema, hemorrhage and cell death. Astrocytes in culture exposed to interleukin-1β cease producing the gap junction protein, Cx43, and begin to produce claudin-1 (Duffy et al, 2000). Claudin promotes the activation of MMP-2, which would allow for amplification to take place, increasing the extent of the injury to the BBB (Miyamori et al, 2001).
An important observation of this study is the ability of a broad spectrum MMP inhibitor to reduce the BBB damage by reducing degradation of the TJPs. A potential role for MMP inhibitors in stroke was recently demonstrated in studies that showed that prevention of the opening of the BBB by MMP inhibitors reduced the toxicity of tissue plasminogen activator in acute stroke (Lapchak et al, 2000; Pfefferkorn and Rosenberg, 2003; Lo et al, 2004). Our results provide a molecular basis for the action of MMP inhibitors.
In conclusion, our results provide direct evidence that changes in the TJPs, claudin-5, and occludin, are occurring through the action of MMP-2 in the early reversible disruption of the BBB, and that MMP-9 is important in the delayed damage. We showed that an MMP inhibitor could block the fragmentation of the TJPs during the early transient opening of the BBB. However, in the later stages, TJPs appear in the astrocytes around the blood vessels. Whether the astrocytes take up the degraded TJPs or synthesize them in response to the injury will require further study.
Footnotes
Acknowledgements
We thank Justin Tibbitts for technical support with the PCR studies.