
Abstract
Although the blood-brain barrier effects of cerebral ischemia have been extensively examined, less attention has focused on ischemia-induced damage to the choroid plexuses that form the blood-cerebrospinal fluid (CSF) barrier (BSCFB). This study examined the rat lateral ventricle choroid plexuses (LVCP) in three ischemic models, bilateral common carotid artery occlusion (2VO) + hypotension with or without reperfusion and permanent middle cerebral artery (MCA) occlusion with or without a tandem common carotid artery occlusion. Blood flow was assessed using [14C]-N-isopropyl-p-iodoamphetamine, and LVCP injury by tissue edema, alterations in [14C]glutamine transport and BSCFB disruption (measured with [3H]inulin). 2VO + hypotension caused an 87% reduction in LVCP blood flow (P < 0.01) and a progressive reduction in LVCP glutamine transport. In contrast to cortex, there was no LVCP hyperemia or delayed hypoperfusion on reperfusion, but there was marked BSCFB disruption. After 30 mins of 2VO + hypotension with 6 h of reperfusion, the [3H]inulin entry into CSF was increased threefold (P < 0.05). Blood-CSF barrier rather than blood-brain barrier disruption appeared to be the main cause of enhanced [3H]inulin entry into hippocampus. Middle cerebral artery occlusion with and without a tandem common carotid artery occlusion only caused 53% and 38% reductions in LVCP blood flow but induced LVCP edema. Results suggest that the LVCP is selectively vulnerable to ischemic injury in terms of the absolute blood flows or, for the MCA occlusion models, the % reductions in flows required to induce injury. BCSFB disruption early after ischemia may enhance the movement of compounds from blood to areas close to the ventricular system and participate in delayed neuronal death.
Introduction
The movement of solutes between blood and brain is regulated by the presence of barrier tissues at the blood–brain and blood–cerebrospinal fluid interfaces (Laterra et al, 1998). Thus, the endothelial cells of the cerebral capillaries and their linking tight junctions form the blood-brain barrier (BBB), while the choroid plexus (CP) epithelial cells and their linking tight junctions form the blood-cerebrospinal fluid (CSF) barrier (BCSFB). There are no tight junctions linking the cells that form the brain-CSF interface, so that even large molecular weight solutes can diffuse between CSF and brain. Apart from the tight junctions that limit paracellular diffusion, both the BBB and the BCSFB have an array of transporters (and metabolic enzymes) that regulate the movement of solutes across these tissues.
It has long been known that cerebral ischemia disrupts the BBB (reviewed in Betz and Dietrich, 1998). Thus, even large molecular weight compounds that are normally excluded by the BBB can begin to gain access during or after an ischemic event. BBB disruption can result in vasogenic edema. Effects of ischemia on the cerebral endothelium, such as the upregulation of adhesion molecules, also contribute to ischemia-induced inflammation. The effects of cerebral ischemia on the BCSFB have, however, received very little attention (reviewed in Keep et al, 2005). Pulsinelli et al (1982) noted that forebrain ischemia induced by four-vessel occlusion resulted in CP necrosis at 6 h, but that CP morphology returned to normal by 72 h. Early morphologic damage followed by recovery has also been described in another model of forebrain ischemia, bilateral common carotid artery occlusion (2VO) with hypotension (Palm et al, 1995; Johanson et al, 2000) and focal cerebral ischemia can also damage CP (Gillardon et al, 1996; Nagahiro et al, 1994). There is evidence of apoptosis as well as necrosis in those models (Gillardon et al, 1996; Ferrand-Drake and Wieloch, 1999; Pulsinelli et al, 1982; Johanson et al, 2000). The early time course of CP damage compared with that in the hippocampus, where damage is delayed, has led to the suggestion that BCSFB disruption might modulate the delayed hippocampus injury (Johanson et al, 2000; Ferrand-Drake, 2001).
Despite evidence that cerebral ischemia can affect the CP, there are a number of unknowns. What levels (percent or absolute) of blood flow are required to induce CP damage? The CP is highly vascularized, and plexus blood flows are much higher (three- to 10-fold) than those in cerebral cortex (reviewed in Keep et al, 2005). It is also unknown whether the CP shows the hyperemia and hypoperfusion found in brain parenchyma after reperfusion. Similarly, there is surprisingly little information on how ischemia affects BCSFB permeability and whether changes in the permeability of that barrier might affect the movement of compounds from blood into tissues adjacent to the ventricular system (e.g., hippocampus). The current study, therefore, examined the effect of two models of cerebral ischemia on CP and BCSFB function.
Materials and methods
The protocol for these animal studies was approved by the University of Michigan Committee on the Use and Care of Animals. Adult male Sprague-Dawley rats (Charles River Laboratories) weighing 275 to 350 g were used for all experiments. Rats were allowed free access to food and water before the experiment.
The experiments were in five parts. Part one examined what reductions in blood flow occurred in the lateral ventricle choroid plexuses (LVCP) and brain tissues after 10 mins of 2VO with hypotension and how blood flow responded to reperfusion (for 5, 30, or 120 mins). The second part examined the effects of the same model of cerebral ischemia on CP function and BBB function by measuring blood to CP and blood to brain L-[14C]glutamine transport. Glutamine transport at both the CP and the BBB is Na+-dependent and, therefore, inhibited by ischemia-induced changes in intracellular Na+ concentration (Keep and Xiang, 1995; Ennis et al, 1998; Kawai et al, 1999). Rats were either exposed to 10, 30, or 70 mins of permanent 2VO + hypotension or 10 mins of ischemia with 60 mins of reperfusion or 30 mins of ischemia + 40 mins of reperfusion. The third part examined the effects of 10 or 60 mins of 2VO + hypotension with 6 h of reperfusion on the entry of [3H]inulin from blood into CSF and brain to assess BCSFB and BBB disruption.
Part four and five utilized a different model of ischemia, permanent middle cerebral artery (MCA) occlusion. In part four, the effects of these 2 h of MCA occlusion on LVCP and parenchymal blood flow were examined. In part five, the effects of 24 h of occlusion on LVCP water content were determined.
2VO + Hypotension
Rats were anesthetized with isoflurane (5% for initial induction followed by maintenance with 2%) and femoral artery and vein cannulas inserted. The former was used for blood sampling and pressure measurement, while the latter was used for isotope administration. Ischemia was induced as described by Nordstrom and Siesjo (1978). In brief, both common carotid arteries were occluded with suture or vascular clips (for permanent and transient ischemia), respectively. Then, blood (∼6 mL) was withdrawn from the femoral artery to reduce mean arterial pressure from 35 to 40 mm Hg. The ischemic period was ended by both the removal of the common carotid artery clips and the restoration of blood volume with the previously withdrawn blood. Isoflurane anesthesia was maintained throughout the experiments.
Middle Cerebral Artery Occlusion
Occlusion of the MCA was achieved using the suture method of Longa et al (1989). Briefly, the rats were anesthetized with pentobarbital (65 mg/kg; intraperitoneally). The bifurcation of common carotid artery was then exposed and the external carotid artery was ligated distally. A 3-0 monofilament nylon suture, its tip rounded by heating, was introduced into the internal carotid artery lumen through the stump of the external carotid artery and gently advanced into the internal carotid artery 19 to 20 mm past the common carotid artery bifurcation to block the origin of MCA. For blood flow experiments involving 2 h of occlusion, anesthesia was maintained throughout. For the 24 h edema experiments, the animals were allowed to recover after surgery and they were reanesthetized with pentobarbital just before killing.
Cerebral Blood Flow
Cerebral blood flow was measured by the indicator fractionation technique (Van Uitert and Levy, 1978) using of [14C]-N-isopropyl-p-iodoamphetamine ([14C]IMP; American Radiolabeled Chemicals, St Louis, MO, USA; Williams et al, 1991a). This method uses an intravenous bolus injection of a blood flow indicator followed by a constant rate of blood withdrawal through a femoral artery catheter to obtain the integral of the arterial isotope concentration. The withdrawal was started 5 secs before intravenous injection of 5 μCi of [14C]IMP. One minute later, the animal was killed by decapitation and blood withdrawal was stopped. The sample of withdrawn arterial blood was bleached with 30% hydrogen peroxide (H2O2) prior addition of scintillation fluid and counting using a Beckman 3801 liquid scintillation counter. Brain sampling differed dependent on the ischemic model. For the 2VO + hypotension model, bilateral samples of LVCP, anterior cortex, and hippocampus as well as cerebellum were taken. For the MCA occlusion model, the hemispheres were divided and LVCP samples taken. The brain stem, hippocampus, and thalamus were discarded. The remaining telencephalic tissues ipsi- and contralateral to the MCA occlusion were placed flat and the portion inferior to the rhinal fissure was removed. A sample from the core of the MCA territory from each hemisphere was then taken using a 5 mm cork borer (Dickinson and Betz, 1992). All brain tissues were weighed and digested in methylbenzethonium hydroxide before counting. Special care was taken with the LVCP samples (because of their small size), which were dissected and immediately weighed on an electronic analytical balance (model AE 100, Mettler Instrument Co., Highstown, NJ, USA) to obtain the wet weight to 0.01 mg (other samples were weighed to 0.1 mg).
Blood flow rates for the individual pieces of brain tissue were calculated using the following equation:
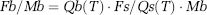
where Fb is the the brain-blood flow; Mb is the the brain mass (in g); Qb(T) is the the quantity of indicator in the tissue at time T; Fs is the rate of blood withdrawal from t = 0 to t = T; and Qs(T) is the quantity of indicator present in the withdrawal at time T. The CBF is expressed as ml/g min.
Blood–Cerebrospinal Fluid and Blood-Brain Barrier Disruption
Blood–CSF and blood-brain barrier disruption was assessed by measurement of the permeability surface (PS) area product for [3H]inulin (New England Nuclear, Boston, MA, USA) using a method modified from Ohno et al (1978) with [14C]inulin (American Radiolabeled Chemicals, St. Louis, MA, USA), circulated for a short period of time, being used to correct for cerebral plasma volume. The [3H]inulin was purified using a G-25M sephadex column (PD-10, Pharmacia) before use. [3H]inulin (80 μCi) was injected through a femoral vein 30 mins before the end of the experiment, while [14C]inulin (15 μCi), being used as a plasma volume marker, was administered as a second injection 2 mins before the end of experiment. The arterial concentration of [3H]inulin was determined using a peristaltic pump (Gilson) to create an artificial organ by continuous withdrawal of blood (0.05 mL/min) into a polyethylene catheter (PE 205). At the end of experiment, terminal CSF (cisterna magna puncture) and plasma samples were taken and the rat was killed by decapitation. Brains were rapidly removed and anterior cortex and hippocampus were sampled as described above. Samples were immediately weighed and digested in methylbenzethonium hydroxide. Scintillation fluid was added to the brain, CSF, and plasma samples, and radioactivity was counted using a Beckman 3801 liquid scintillation counter (Fullerton, CA, USA). The PS area product for [3H]inulin entry into the brain or CSF was calculated from the formula:
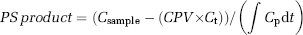
where Csample is the counts per gram of brain or CSF sample, CPV is the plasma volume of the sample determined from the [14C]inulin space, Ct is the terminal plasma concentration of [3H]inulin, and ∫Cpdt is the integral of the [3H]inulin plasma concentration (Cp) for the experiment. The latter was calculated from the radioisotope content of the continuously withdrawn arterial blood sample.
Glutamine Transport into Brain and Choroid Plexus
The transport of L-[14C]glutamine (Amersham Biosciences, Piscataway, NJ, USA) into the brain and LVCP was assessed by measurement of the PS product as described previously (Kawai et al, 1999). In brief, a bolus of 0.2 mL saline containing 5 μCi L-[14C]glutamine was administered 5 min before decapitation of rats. After radioisotope injection, an arterial blood sample was continuously withdrawn at a constant rate to determine the integral of plasma radioactivity. For the simultaneous determination of plasma volume, 20 μCi of [3H]inulin was then injected into the femoral vein and allowed to circulate for 2 mins before decapitation. Tissue processing and calculation of the L-[14C]glutamine PS product were as for measurement of the inulin PS product.
Measurement of Choroid Plexus Water Content
Rats were decapitated at 24 h after MCA occlusion under pentobarbital anesthesia (65 mg/kg). The LVCP were dissected and immediately weighed on an electronic analytical balance (model AE 100, Mettler Instrument Co.) to obtain the wet weight (to 0.01 mg). They were then dried in an oven at 100°C for 24 h to obtain the dry weight and the water content determined as (wet weight—dry weight)/dry weight.
Statistical Analysis
All data in this study are presented as mean ± s.d. Data were analyzed with Student's t-test, except when multiple comparisons were made to a control when ANOVA followed by a Newman–Keuls test was used. Data examining the relation between [3H]inulin entry into the brain and CSF were analyzed by least-squares linear regression. Statistical significance was accepted at P < 0.05.
Results
Two-Vessel Occlusion + Hypotension
Blood flows in the LVCP of control rats were 2.5 ± 0.4 mL/g min, similar to that found in the fourth ventricle CP (2.7 ± 0.5 mL/g min). The CP values were approximately threefold greater than found in the anterior cerebral cortex and hippocampus (0.94 ± 0.15 and 0.92 ± 0.11 mL/gmin). After 10 mins of 2VO with hypotension, the LVCP blood flows were markedly reduced (P < 0.01) to 13% of control values (Figure 1B). By comparison, anterior cortex and hippocampus blood flows were reduced to 7% and 21% of control values, respectively (Figure 1A). The cerebellum, brain stem, and fourth ventricle CP blood flows were all ∼70% to 80% of control values.
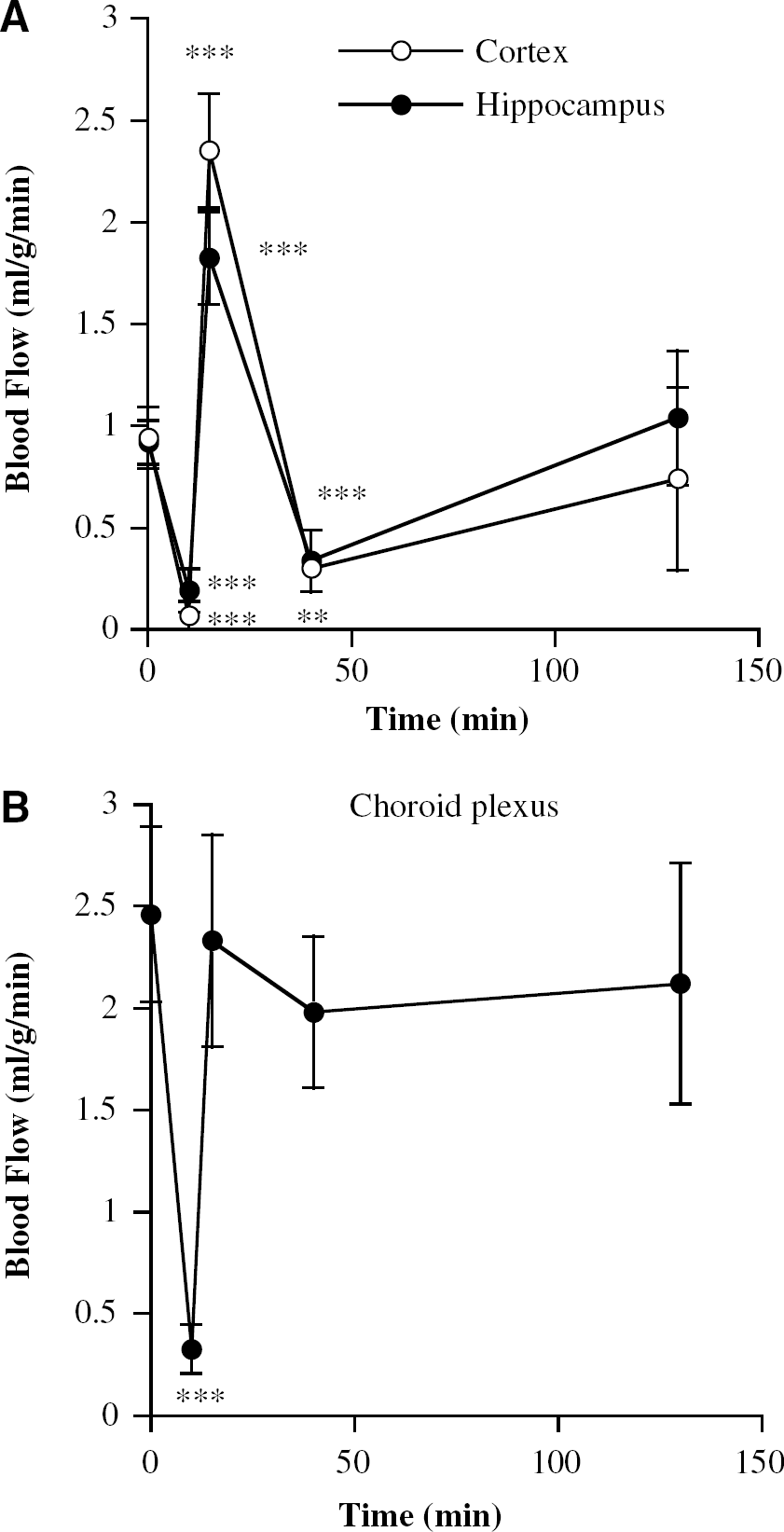
Blood flows in (
As expected, after 5 mins of reperfusion there was hyperemia in the anterior cortex and hippocampus (Figure 1A). This was followed by hypoperfusion after 30 mins of reperfusion and a return towards normal by 120 mins (Figure 1A). By contrast, in the LVCP, blood flows returned to approximately control levels on reperfusion and did not significantly change over the next 2 h (Figure 1B). Thus, we found no evidence of hyperemia or hypoperfusion during reperfusion of the LVCP.
To examine whether the reductions in LVCP blood flow were sufficient to affect function, the uptake of L-[14C]glutamine from blood to the CP (and brain) was examined. Glutamine uptake into the CP and brain is via a secondarily active process (Na-dependent; Keep and Xiang, 1995; Ennis et al, 1998). With a glutamine uptake measured over the last 5 mins of the occlusion, 10 mins of 2VO + hypotension caused a ∼45% reduction in L-[14C]glutamine uptake into the LVCP compared with 61% and 32% reductions in uptake into anterior cortex and hippocampus, respectively (Figure 2). Prolonging the ischemia for 30 mins resulted in an even greater decline in all three tissues (72%, 86%, and 63% reductions in the LVCP, the anterior cortex, and the hippocampus, respectively; Figure 2), although there was no further significant decrease with 70 mins of permanent ischemia.
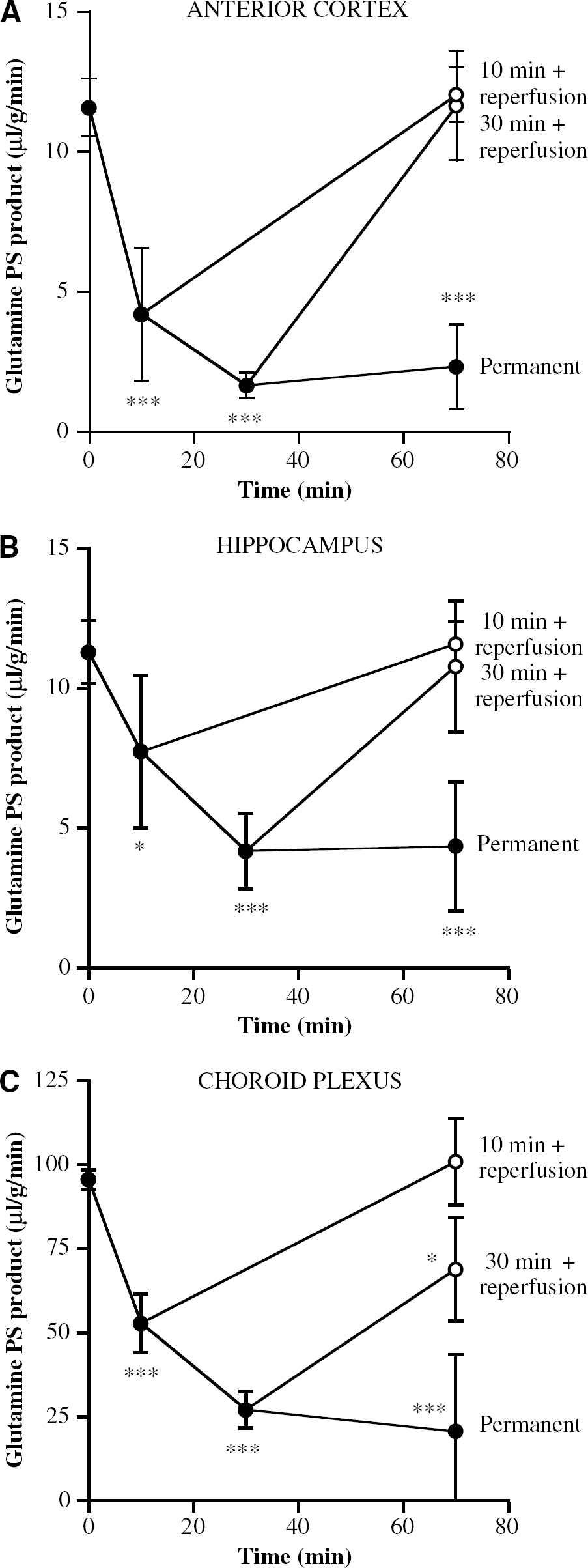
Blood to tissue [14C]glutamine transport (permeability surface area (PS) product) in (
Reperfusion after 10 mins of 2VO + hypotension resulted in a normalization of L-[14C]glutamine transport from blood to the LVCP, anterior cerebral cortex, and hippocampus (Figure 2). Even when the ischemia was prolonged to 30 mins, blood to the anterior cerebral cortex and hippocampus returned to preischemic values (Figure 2). Blood to LVCP glutamine transport, however, remained depressed (Figure 2).
Blood-CSF barrier permeability was assessed with [3H]inulin. In control animals, the PS product for [3H]inulin entry into CSF was approximately three- to fourfold greater than for entry into the anterior cortex or hippocampus. Six hours after 10 mins of 2VO + hypotension, BCSFB permeability was doubled, whereas it was trebled after 30 mins of 2VO + hypotension with 6 h of reperfusion (Figure 3A). Interestingly, despite the fact that 2VO + hypotension caused more severe ischemia in the anterior cortex than in the hippocampus (Figure 1), the effect of ischemia + reperfusion on the [3H]inulin PS product was actually greater in the hippocampus. As with the BCSFB, the hippocampus [3H]inulin PS product was approximately trebled after 30 mins of 2VO + hypotension with 6 h of reperfusion (Figure 3A). Indeed, there was a good correlation between hippocampus and CSF PS products (Figure 3B).
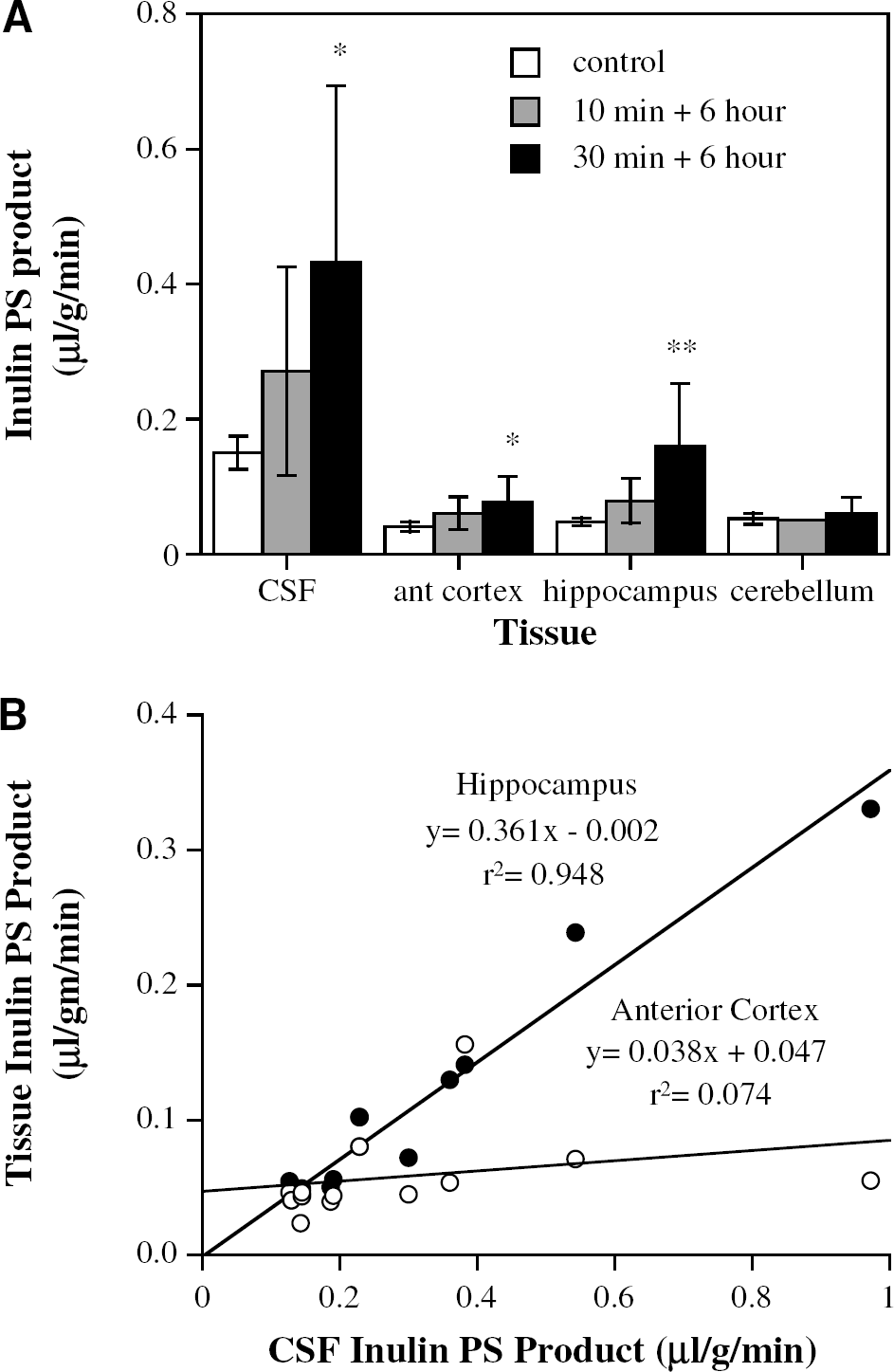
(
Middle Cerebral Artery Occlusion
As with the ipsilateral cortex, there was a significant reduction in blood flow to the ipsilateral LVCP after 2 h of MCA occlusion with or without a tandem carotid artery occlusion (Figure 4). The blood flows in the ipsilateral LVCP were 0.75 ± 0.33 and 1.06 ± 0.44 mL/gmin with or without a tandem carotid artery occlusion, respectively, compared with ∼1.75 mL/gmin in the contralateral LVCP. Although these ischemic models only resulted in a 53% reduction in LVCP blood flow with the tandem occlusion and a 38% reduction in flow with no tandem occlusion, both models resulted in significant CP edema after 24 h of permanent ischemia. Thus, with the tandem occlusion, the water content of the ipsilateral LVCP was 4.82 ± 0.67 g/g dry weight compared with 3.56 ± 0.26 g/g dry weight in the contralateral LVCP (P < 0.05; n = 4/group). With MCA occlusion without the tandem occlusion, water contents of the ipsi- and contralateral LVCP were 4.72 ± 0.72 and 3.84 ± 0.57 g/g dry weight, respectively (P < 0.05; n = 6/group).
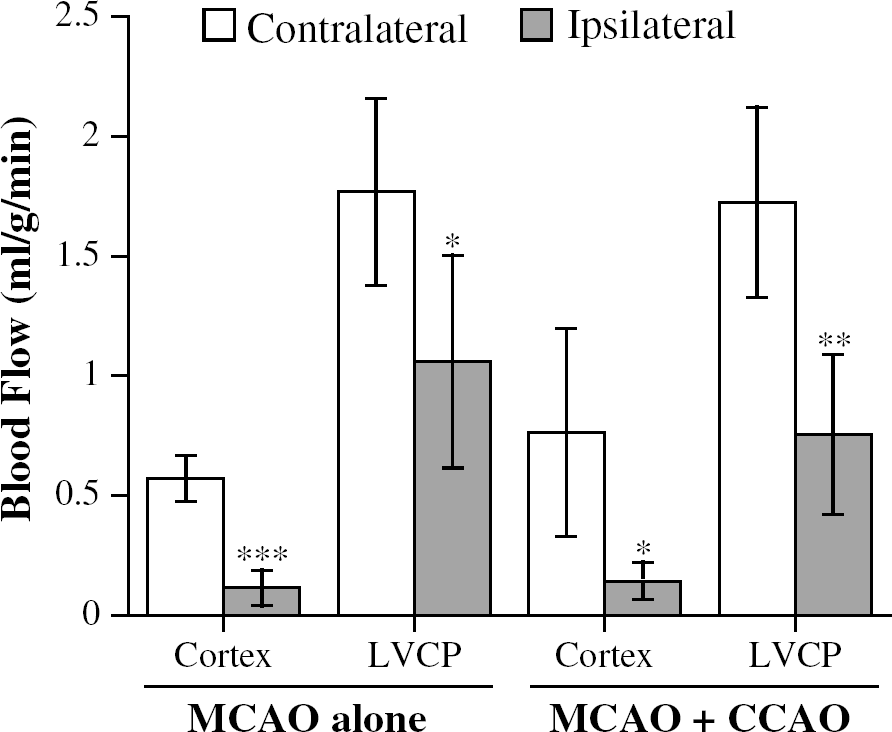
Blood flow in the ipsi- and contralateral cortex (core of middle cerebral artery (MCA) territory) and in the ipsi- and contralateral lateral ventricle choroid plexuses (LVCP) 2 h after permanent MCA occlusion. There were two groups of animals, those with a tandem common carotid artery occlusion (MCAO + CCAO) and those with just the MCA occlusion (MCAO alone). Values are means ± s.d., n = 5, *, ** and ***Significant differences from contralateral at the P < 0.05, P < 0.01 and P < 0.001 levels, respectively.
Discussion
The purpose of this study was to quantify changes in CP blood flow during and after cerebral ischemia to elucidate the vulnerability of this tissue to ischemic injury and to determine the magnitude of BCSFB disruption and whether that disruption might influence nearby brain tissue. In all three models tested (2VO + hypotension, and MCA occlusion ± tandem carotid artery occlusion), there was evidence of CP injury during ischemia even though the absolute CP ischemic blood flows were higher than the threshold that induces injury in brain parenchyma, suggesting selective CP vulnerability. Similarly, in the 2VO + hypotension model, there was evidence that BCSFB disruption occurs before damage to the BBB. Tissues adjacent to the ventricular CSF (such as hippocampus) appear, therefore, to be exposed to higher concentrations of blood-borne solutes than areas distant from the ventricles. Interestingly, despite evidence of CP/BCSFB damage, there was no evidence of hyperemia/delayed hypoperfusion on reperfusion in the 2VO + hypotension model.
Morphologic and Methodological Considerations
In rat, the lateral ventricle CP blood supply is from the anterior and posterior choroidal arteries. As in man, the rat anterior choroidal artery is usually a branch off the internal carotid artery. He et al (2000) found that the branching is situated 0.5 to 2 mm proximal to MCA bifurcation from the internal carotid artery. The posterior choroidal artery is a branch of the posterior cerebral artery (Hogendorf et al, 2001). The dual blood supply to the LVCPs probably offers some protection from focal ischemic events (e.g., blockage to one of the choroidal arteries), although there is evidence in man that a blockage in the posterior choroidal artery can induce CP infarction (Liebeskind and Hurst, 2004).
The first part of this study focused on the 2VO + hypotension model of ischemia for two main reasons. First, there are morphologic studies indicating that the LVCPs are affected by this model (Palm et al, 1995; Johanson et al, 2000). Second, this is a bilateral model. In the BCSFB disruption experiments, sampled CSF (from the cisterna magna) will reflect entry of [3H]inulin across the lateral and the fourth ventricle CP. Any enhanced influx across a damaged CP will be diluted by normal influx across undamaged CP. By affecting both LVCP, 2VO + hypotension model enhances the possibility of detecting changes in permeability in the CSF sample. In the rat, the two LVCP combined weigh approximately the same as the fourth ventricle CP (Quay, 1972). Assuming that the CSF sample reflects equally the lateral and fourth ventricle CP (the latter plexus is unaffected by the ischemia), this suggests that the threefold increase in the PS product for inulin entry into CSF after 6 h of reperfusion would actually reflect a fivefold increase in LVCP permeability.
The second part of the study examined the thread model of MCA occlusion with and without a tandem common carotid artery occlusion. These models were chosen because (a) they are very common models of cerebral ischemia, (b) there is previous evidence of LVCP damage after MCA occlusion (Gillardon et al, 1996; Nagahiro et al, 1994), (c) a reduction in CP blood flow might be expected in these models because the internal carotid artery suture occluding the MCA bifurcation may also occlude the anterior choroidal artery, and (d) we expected reduction in CP blood flow would be less than that found in the 2VO + hypotension model, allowing examination of graded decreases in blood flow.
Four end points were examined, cerebral blood flow, glutamine transport, BCSF barrier disruption, and CP edema. Blood flow was measured with [14C]IMP rather than iodo-antipyrine, the tracer most commonly used for brain-blood flow studies. [14C]-N-isopropyl-p-iodoamphetamine has the advantage that on entry across the vasculature, it remains within the tissue whereas iodo-antipyrine may back flux to blood or, in the case of the CP, efflux to CSF (Williams et al, 1991a, b). [14C]-N-isopropyl-p-iodoamphetamine has been used to examine control CP blood flow previously (Williams et al, 1991a; Szmydynger-Chodobska et al, 1994) and the blood flows obtained with [14C]IMP are similar to those obtained in other species by other techniques (reviewed in Williams et al, 1991a). [14C]-N-isopropyl-p-iodoamphetamine is almost completely extracted by the brain during a single pass (Kuhl et al, 1982) and it can be used to assess blood flow in even highly perfused areas of the brain such as the control CP (Williams et al, 1991a). Once within the tissue, it becomes trapped (acting as a fluid microsphere; Szmydynger-Chodobska et al, 1994). The effects of ischemia on this entrapment have not been examined, but two factors suggest that the entrapment is not changed by ischemia. First, the ischemic blood flows determined in brain parenchyma with IMP are similar to those with other markers such as iodoantipyrine. Second, reperfusion of the CP results in a return of blood flow to normal; that is, any change in entrapment during ischemia would have to be transient. It should also be noted that any reduced entrapment during ischemia and a potential reduced extraction at low pH would result in an underestimate of CP blood flow. In terms of data interpretation, this would not affect, but rather enhance, our conclusion that relatively small reductions in CP blood flow can cause CP damage.
One of the earliest effects of cerebral ischemia is a redistribution of ions between the intra- and extracellular compartments (Kristian and Siesjo, 1997). Increases in intracellular Na+ in CP epithelial cells and cerebral endothelial cells will decrease [14C]glutamine transport since such transport at the CP and the BBB is Na+-dependent (Keep and Xiang, 1995; Ennis et al, 1998). The use of this marker allowed assessment of CP injury during the short ischemic period as well as during reperfusion. Palm et al (1995) have reported an increase in CP Na+ after 2VO + hypotension with reperfusion. An increase in tissue Na+ can also cause a concomitant increase in tissue water (Palm et al, 1995) and edema was used as a marker of CP injury in our MCA occlusion experiments.
To examine BCSFB, the PS product for [3H]inulin was measured. The PS products for the compound at the normal BCSFB and BBB are low (∼0.15 and 0.05 μL/gmin, respectively), facilitating measurement of disrupted barrier function. In addition, the molecular weight of inulin (∼5,000) is similar to a number of peptides that might enter the brain from blood when either the BSCFB or BBB are disrupted.
Effects of Ischemia on the Choroid Plexus
In each of the ischemia models used in this study, there was evidence of CP dysfunction. Thus, during 2VO + hypotension there is a marked, time-dependent, reduction in blood to CP [14C]glutamine transport that is similar to the reduction in transport found at the BBB. During reperfusion in that model, there is a restoration of transport, although it appears to be incomplete if the ischemic event is 30 rather than 10 mins long. This model of ischemia also resulted in a significant degree of BCSFB disruption. Thus, 10 and 30 mins of ischemia with 6 h of reperfusion resulted in an approximate doubling and trebling of the PS product for [3H] inulin entry into CSF, respectively. As noted above, these measures underestimate the effect of the ischemia on the LVCP, since the CSF sampled reflects entry across the LVCP and the fourth CP. In the permanent MCA occlusion models, injury was evident by CP edema.
The current study did not examine the effects of ischemia on CP cell death. However, studies have shown profound morphologic changes and necrosis in the LVCP epithelium in the 2VO + hypotension and the four vessel models of forebrain ischemia (Johanson et al, 2000; Pulsinelli et al, 1982). In those studies, tissue injury peaked early in reperfusion (∼6 h) and was reduced at 24 h. DNA fragmentation in the LVCP also occurs with 2VO + hypotension and with permanent MCA occlusion (Ferrand-Drake and Wieloch, 1999; Gillardon et al, 1996).
There were ∼58% and 40% reductions in LCVP blood flows in the MCA occlusion model with and without tandem occlusion and an 87% reduction with 2VO + hypotension. In brain parenchyma, thresholds have been defined describing the relation between cerebral blood flow and tissue dysfunction. Thus, reductions in blood flow below ∼0.19 mL/g min in the rat will cause cortical energy failure (Mies et al, 1991). As a % decline, this is about 80% reduction in flow. For the LVCP, smaller % declines in blood flow can cause damage. Middle cerebral artery occlusion resulting in a 40% reduction in LVCP blood flow causes LVCP edema (this study) and apoptosis (Gillardon et al, 1996) as well as BCSFB disruption (Nagahiro et al, 1994). In terms of absolute blood flows, the more severe 2VO + hypotension model resulted in LVCP flows of 0.33 ± 0.11 mL/g min. This is above the threshold for cerebral cortex energy failure, but it causes LVCP necrosis, DNA fragmentation, profound morphologic changes, and marked disruptions in transport and BCSFB integrity (Palm et al, 1995; Johanson et al, 2000; Ferrand-Drake and Wieloch, 1999; and results reported above). In total, this suggests that the CP is selectively vulnerable to ischemic damage. This is supported by the early work of Pulsinelli et al (1982) who found that forebrain ischemia caused much earlier damage to the LVCP than the hippocampus.
It is uncertain why the CP should be selectively vulnerable to ischemia. However, it should be noted that the CP epithelium has an apparent high metabolic demand as evinced by a high mitochondrial density (at about 7.6% of cell volume, about 2% greater than cortical tissue; Keep and Jones, 1990a, b). In addition, these are nonexcitable cells. In neurons, a reduction in metabolism will impact activity before inducing cell injury. In the CP epithelium, there may be relatively few nonessential functions that can be shutdown to preserve tissue integrity. The dual blood supply from the anterior and posterior choroidal arteries may be a reflection of this vulnerability since a dual supply would aid in preventing reductions in LVCP blood flow.
As expected, there was hyperemia in the hippocampus and anterior cortex on reperfusion after 10 mins of 2VO + hypotension. In marked contrast, there was no hyperemia in the LVCP. Evidence indicates that parenchymal hyperemia after cerebral ischemia is because of the build up of metabolites during the ischemic events that lead to maximal vasodilatation (Dirnagl and Lindauer, 1998). For the CP, it is possible that the CSF acts as a sink preventing the build up of metabolites that could cause hyperemia. Thus, although hyperemia may participate in early BBB disruption after ischemia (Kuroiwa et al, 1985), this is not the case with BCSFB disruption.
Similarly, although there was delayed hypoperfusion in both cortex and hippocampus, there was no delayed hypoperfusion in the LVCP in the 2VO + hypotension. In parenchymal tissues, delayed hypoperfusion has been attributed to ischemia-induced alterations in arteriolar tone (changed vasoconstrictor/vasodilator release; Hauck et al, 2004). It is possible that the high absolute blood flow rate to the CP protects the arterioles from damage during ischemia. It is also notable that CP endothelial cells have a lower mitochondrial content than parenchymal cerebral blood vessels (Keep and Jones, 1990a, b) and, thus, potentially a lower metabolic rate. This may limit CP endothelial damage during ischemia and prevent delayed hypoperfusion.
Impact of Blood–Cerebrospinal Fluid Barrier Dysfunction on Parenchyma
The proximity of different brain tissues to the CSF system differs. Thus, the rat anterior cortex has been chosen previously as a sample distant from the ventricular system and, therefore, less influenced by entry of compounds across the LVCP (Smith and Rapoport, 1986). In contrast, the hippocampus is in close proximity to the LVCP and the ventricular CSF. In our results, 2VO + hypotension caused much greater increases in the entry of [3H]inulin into the hippocampus than into the anterior cortex (three- to fourfold greater after 30 mins ischemia and 6 h of reperfusion). This occurred even though ischemic blood flows were lower in the anterior cortex (7% of normal) than in hippocampus (21% of normal). This suggests that the increased [3H]inulin entry into hippocampus is because of disruption of the adjacent BCSFB rather than disruption of the tissues own BBB. This suggestion is supported by the fact that there is a good correlation between increases in the entry of [3H]inulin into CSF and hippocampus (in contrast to the correlation between CSF and anterior cortex).
This study does not address the question of whether BCSFB disruption has a detrimental or beneficial effect on tissues adjacent to the ventricular system. It has been suggested that the entry of potentially toxic compounds across a disrupted BCSFB might participate in delayed neuronal injury in the hippocampus (Ferrand-Drake, 2001). It has also been suggested that enhanced influx of growth factors across the BSCFB might serve to limit injury (Johanson et al, 2000). It should also be noted that the CP produces growth factors itself that may limit ischemic injury in the brain (Johanson et al, 2003), although that will depend on the extent to which production is not prevented by CP damage. Determining the importance of damage to the CP/BCSFB system really requires a method of preventing CP damage and then examining the effect of such a method on hippocampus injury.
Ischemia-induced BCSFB disruption may also be important in terms of drug delivery to the brain in those strokes involving reductions in LVCP blood flow (i.e., affecting the anterior and/or posterior choroidal arteries). The earlier disruption of the BCSFB compared with the BBB is an important consideration because of the time windows for potential therapies for stroke.
In conclusion, this study provides evidence for CP (BCSF barrier) injury in three models of stroke. The CP appears to be selectively vulnerable to ischemia, although this tissue does not experience hyperemia/hypoperfusion on reperfusion. Choroid plexus injury during ischemia may have a significant impact on tissues that are adjacent to the ventricular system.