
Abstract
Prostacyclin (PGI2), a potent vasodilator and inhibitor of platelet aggregation and leukocyte activation, is crucial in vascular diseases such as stroke. Prostacyclin synthase (PGIS) is the key enzyme for PGI2 synthesis. Although expression of PGIS was noted in the brain, its role in ischemic insult remains unclear. Here we reported the temporal and spatial expression of PGIS mRNA and protein after 60-min transient ischemia. Northern blot and in situ hybridization revealed a delayed increase of PGIS mRNA in the ischemic cortex at 24- to 72-h after ischemia; PGIS was detected mainly in the ipsilateral penumbra area, pyriform cortex, hippocampus, and leptomeninges. Western blot and immunohistochemical analysis revealed that PGIS proteins were expressed temporally and spatially similar to PGIS mRNA. PGIS was heavily colocalized with PECAM-1 to endothelial cells at the leptomeninges, large and small vessels, and localized to neuronal cells, largely at the penumbra area. A substantial amount of PGIS was also detected in the macrophage and glial cells. To evaluate its role against ischemic infarct, we overexpressed PGIS by adenoviral gene transfer. When infused 72 h before ischemia (–72 h), Adv-PGIS reduced infarct volume by ~50%. However, it had no effect on infarct volume when infused immediately after ischemia (0 h). Eicosanoid analysis revealed selective elevation of PGI2 at −72 h while PGI2 and TXB2 were both elevated at 0 h, altering the PGI2/thromboxane A2 (TXA2) ratio from 10 to 4. These findings indicate that PGIS protects the brain by enhancing PGI2 synthesis and creating a favorable PGI2/TXA2 ratio.
Introduction
In cerebral ischemia, Ca2+ overload led to the activation of phospholipases and the subsequent generation of arachidonic acid (AA), which is rapidly converted to prostaglandins (PGs), thromboxane A2 (TXA2) and leukotrienes (LTs) (Bazan, 1970; Wolfe, 1982; Chen et al, 1986). These metabolites contribute to the irreversible neuronal damage (Chan and Fishman, 1978; Chan et al, 1983; Hsu et al, 1989). Among the eicosanoids substance, prostacyclin (or PGI2) is a potent vasodilator and inhibitor of platelet aggregation, leukocyte activation, and leukocyte–endothelial interactions (Moncada, 1983). Prostacyclin antagonizes the proaggregation and vasoconstrictive actions of TXA2. A PGI2/TXA2 imbalance is detrimental and can lead to vascular diseases such as stroke (de Levai et al, 2004). It has been shown that intravenous PGI2 administration prevented ischemic brain damage in experimental studies (Matsuda et al, 1997), and stable PGI2 analogs administered directly into brain ventricles exert neuroprotective actions by increasing blood flow, reducing platelet aggregation, and protecting neurons (Cui et al, 1999; Takamatsu et al, 2002). However, controlled clinical trials have not confirmed the beneficial effect of PGI2 (Martin et al, 1985; Bath, 2004).
Prostacyclin synthesis is catalyzed by three successive enzymatic reactions: (1) liberation of AA from membrane phospholipids by activated cytosolic phospholipase A2; (2) conversion of AA to PGH2 by cyclooxygenase (COX or PGH2 synthase); and (3) conversion of PGH2 to PGI2 by PGI2 synthase (PGIS). Cyclooxygenase is the rate-limiting step for PGs synthesis; two distinct isozymes encoded by separate genes have been identified (Simmons et al, 2004). Cyclooxygenase-1 is constitutively expressed and plays an important physiologic role, whereas COX-2 is inducible and plays diverse physiologic and pathophysiologic roles. Human PGIS gene is about 60 kb long and contains 10 exons, which was assigned to chromosome 20. Prostacyclin synthase is a heme-containing protein belonging to the family of cytochrome P450. It is an atypical cytochrome P450 as it does not possess monooxygenase activity (de Levai et al, 2004). Prostacyclin synthase catalyzes the isomerization of PGH2 to form PGI2. Prostacyclin synthase undergoes suicidal inactivation during catalysis, which limits the extent of PGI2 production (Sanduja et al, 1994; Wang and Chen, 1996).
Prostacyclin synthase is expressed in the brain of rat, bovine, and human (Mehl et al, 1999). Association of PGIS gene variant with cerebral infarction has been reported (Nakayama et al, 2000). Prostacyclin synthase-null mice developed ischemic disorders (Yokoyama et al, 2002). Bicistronic COX-1/PGIS gene transfer is capable of reducing infarct volume after brain ischemia–reperfusion injury (Lin et al, 2002a). However, it is unclear whether PGIS expression is altered by brain ischemia. To the best of our knowledge, the temporal and spatial expression profiles of PGIS mRNA and protein in the ischemic brain has not been previously reported. The present study is undertaken to determine the temporal and spatial expression of PGIS after focal cerebral ischemia–reperfusion, and the role of PGIS gene transfer in selective augmentation of PGI2 production and neuroprotection in a well-established rat middle cerebral artery occlusion (MCAO) model.
Materials and methods
Stroke Model
The rat focal cerebral ischemia–reperfusion model was described previously (Lin et al, 1993, 2002b). In brief, right middle cerebral artery (MCA) of male Long-Evans rats was reversibly ligated under a stereomicroscope. Both common carotid arteries were then occluded using nontraumatic aneurysm clips. After 60 mins of ischemia, arterial occlusion was released. In this model, ischemia for 60 mins produced a large infarct confined to the right MCA cortex region with ~90% regional blood flow reduction (Lin et al, 2003). While under anesthesia, the rectal temperature was monitored and maintained at 37.0°C ± 0.5°C by using a homeothermic blanket (Harvard, Holliston, MA, USA). After the ischemic insult, rats were kept in an air-ventilated incubator at 24.0°C ± 0.5°C. Arterial blood gases, mean arterial pressure, and heart rate were also monitored in selected animals before and during ischemia and for 30 mins after the initiation of reperfusion. The values were within normal range before, during, and after ischemia. At the end of each experiment, rats were either killed by decapitation under anesthesia, and the brains quickly removed to collect the cerebral cortex, or killed by transcardial perfusion with normal saline under anesthesia followed by cold 4% paraformaldehyde. The whole brains were removed and cryoprotected in 30% sucrose at 4°C overnight. All the procedures were in accordance with the Public Health Service Guide Approved Procedures for the Care and Use of Laboratory Animals and approved by the Academia Sinica Institutional Animal Care and Utilization Committee (AS IACUC).
RT-PCR and Northern Blot Analysis
Total RNA was isolated from the cerebral cortex and subjected to RT-PCR as previously described (Lin et al, 2000). Polymerase chain reaction (PCR) primers for PGIS were chosen from sequences in the Genbank: PGISF, 5'-GCC,ATC,TTC,CTC,ATG,GAG,AGG,ATT,TTT,GAT-3';PGISR, 5'-ACC,CAT,ATT,CCC,CTG,TGT,GGC,CCA,CAG,CTG-3'. The expected length of the PCR product is 600 bp. PCR was performed in a thermal controller for 30 cycles using the following profile: (1) 94°C for 7 mins; (2) repeat cycles at 94°C for 30 sees, 55°C for 30 sees, 72°C for 1 min; and (3) 72°C for 10 mins (GeneAmp 2400, PE). The size of the PCR product was verified on 2% agarose gel electrophoresis. The product was eluted and subcloned (Invitrogen, CH Groningen, The Netherlands) for sequence identity, which served as a probe to detect PGIS mRNA in Northern blot analysis and in situ hybridization.
Northern blot analysis has been described previously (Lin et al, 2000). Briefly, RNA samples (15 µg/lane) were applied to 1.2% agarose gel in the presence of 2.2 mol/L formaldehyde. After electrophoresis, gels were transblotted onto Nytran membranes (Gene Screen Plus, DuPont, Boston, MA, USA). Membranes were prehybridized at 60°C in a solution containing 1% SDS, 1 mol/L NaCl, 10% dextran sulfate, and 100 µg/mL sheared salmon sperm DNA. RT-PCR amplified PGIS and GAPDH cDNA probes were labeled with 32P-dCTP using the randomprimer labeling method (Amersham, Arlington Heights, IL, USA). Radioactive probes (1 × 106 cpm/mL) were added directly to the prehybridization solution. After overnight hybridization at 60°C, membranes were washed twice in 2 × SSC at room temperature for 5 mins each, followed by two 30-min washes at 60°C in 2 × SSC/1% SDS, and two 30-min washes at 60°C in 0.1 × SSC. Membranes were then exposed to Kodak X-Omat/XB-1 films. The radioactive bands were quantified by a densitometer.
In Situ Hybridization
In situ hybridization to detect the regional distribution of mRNA signals has been described previously (Lin et al, 2003). In brief, brain slices of 25-µm were mounted on poly-
Western Blot Analysis
Analysis of PGIS protein in the cortex was performed as described previously (Lin et al, 2002a), by a specific PGIS antibody kindly provided by Dr K-H Ruan (University of Texas–Houston Medical School). In brief, ischemic cortex was homogenized in lysis buffer (0.32 mol/L sucrose, 1 mmol/L EDTA, 50 mmol/L Tris-Cl pH 7.4, and protease inhibitor cocktail (Roche, Mannheim, Germany)). An equal amount of proteins (20 µg) was applied to SDS-polyacrylamide gels and electrophoresed under the 10% reducing gel condition. Separated proteins were electroblotted onto Hybond-P:PVDF membranes (Amersham). The membranes were blocked in TBST buffer containing 20 mmol/L Tris-HCl, 5% nonfat milk, 150 mmol/L NaCl, and 0.05% Tween 20, pH 7.5, for 1 h at room temperature. The blots were incubated with PGIS antibody (1:1,000), followed by a secondary alkaline phosphatase-conjugated antibody (1:5,000; Promega, Madison, WI, USA). Protein bands were visualized by an enhanced chemiluminescence system (PIERCE, Rockfold, IL, USA).
Immunohistochemical Staining
Immunohistochemical methods to study the regional distribution of gene products have been described previously (Lin et al, 2003). Briefly, brain slices of 25-µm thickness were permeabilized with 0.3% Triton X-100 and incubated overnight at 4°C with a rabbit anti-PGIS antibody (1:100), followed by a biotin-labeled goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA, USA). After washing, the sections were further incubated with ABC Elite complex (Vector Laboratories) and visualized with DAB. Slides were washed, dehydrated, cleared in xylene, and mounted.
For confocal study, brain sections were permeabilized with 0.3% Triton X-100. Rabbit anti-PGIS antibody (1:100) was incubated with one of the following mouse antibodies against PECAM-1 (endothelial marker; 1:100; Chemicon, Temeala, CA, USA), NeuN (neuronal marker; 1:100; Chemicon), Ox-42 (macrophage marker; 1:100; Chemicon), or GFAP (glial marker; 1:40; Boehringer Mannheim, Indianapolis, IN, USA) at 4°C overnight. The sections were then incubated with a Cy™2-conjugated goat anti-rabbit IgG antibody (1:1,000; Jackson ImmunoResearch, West Grove, PA, USA) and Cy™3-conjugated goat anti-mouse antibody (1:1,000; Jackson ImmunoResearch). Slides were washed, wet-mounted, and examined under confocal microscopy.
Preparation of Replication-Defective Recombinant Adenoviral Vectors
The procedure was previously described (Lin et al, 2002a). We constructed in the replication-defective recombinant adenoviral (rAd) vector a human phosphoglycerate kinase (hPGK) promoter to drive PGIS (Adv.hPGK-PGIS) and an hPGK alone to serve as control (Adv.hPGK). Replication-defective rAd vectors were generated by homologous recombination and amplified in HEK 293 cells. Recombinant adenoviral stocks were prepared by CsCl gradient centrifugation, aliquoted, and stored at −80°C. Viral titers were determined by a plaque-assay method. HEK 293 cells were infected with serially diluted viral preparations and then overlaid with low meltingpoint agarose after infection. Numbers of plaques formed were counted within 2 weeks.
Intraventricular Infusion of Adenoviral Constructs
Adv-PGK-PGIS at 5 × 107 plaque-forming units (PFU)/10 µL aCSF were intraventricularly infused as previously reported (Lin et al, 2002a). Anesthetized rats were placed in a stereotaxic apparatus, and 10 µL of rAd was infused into the right lateral ventricle at a rate of 5 µL/min at the following coordinates: A, 2.5 mm caudal to bregma; R, 2.8 mm lateral to midline; and V, 3.0 mm ventral to dural surface. Periodic confirmation of proper placement of the needle was performed with infusion of fast green. To ascertain the transgene expression at the region of rAd infusion, we infused recombinant adenoviruses containing a green fluorescent protein (GFP) gene into the right lateral ventricle and examined the GFP expression in 8 coronal slices of right cortex 72 h later. Green fluorescent protein was visualized in the lining ependymal cells and cells surrounding the right ventricular region in all eight coronal brain slices. Green fluorescent protein was not visualized in the left ventricle. These results confirm the uptake and expression of rAd by ependymal and surrounding cells.
Measurements of Brain Tissue Eicosanoids by Enzyme Immunoassay
Cortex was homogenized gently in 1 mL ice-cold buffer (0.05 mol/L Tris at pH 7.0, 0.1 mol/L NaCl, 0.02 mol/L EDTA) and centrifuged at 55,000 g for 1 h. The supernatant was acidified and passed through a Sep-Pak C18 cartridge. Eicosanoids were eluted with 100% methanol, dried under nitrogen gas, redissolved in a small amount of buffer, and analyzed using enzyme immunoassay kits: PGE2, 6-keto-PGF1α, TXB2, and LTB4 kits from R&D System Ins (Minneapolis, MN, USA) and PGD2 and LTC4 kits from Cayman Chemical Co, (Ann Arbor, MI, USA) (Lin et al, 2002a).
Measurement of Infarct Volume
The infarct volume in the right MCA territory was measured as described (Lin et al, 1993). Rats were euthanized, and the cortex was carefully removed, cooled in ice-cold saline for 5 mins, and dissected coronally into 2-mm slices using a Jacobowitz brain sheer (Zivic-Miller, Allison Park, PA, USA), which were incubated in PBS (pH 7.4) containing 2% 2,3,5-triphenyltetrazolium chloride at 37°C for 30 mins and then stored in 10% neutral-buffered formalin. The cross-sectional area of infarction in the right MCA territory for each brain slice was measured with a Zeiss IBAS image analyzer.
Statistical Analysis
ANOVA was used to compare the temporal expression of infarct volume and levels of eicosanoid. The level of differences among groups was analyzed by Fisher's protected t-tests (GB-STAT 5.0.4, Dynamic Microsystem Inc., Silver Spring, MD, USA). P < 0.05 was considered statistically significant.
Results
The temporal expression profile of PGIS mRNA after ischemia–reperfusion was examined by Northern blot analysis using the PCR product as the probe (Figure 1). A faint ~ 2.1 kb mRNA band was detected in sham control (Figure 1A). Compared with sham-operated controls, PGIS mRNA level in ischemic cortex was not significantly changed until at 24 h when it was increased by approximately 3-fold, which started to decline at 72 h and returned to basal level at 168 h. Prostacyclin synthase mRNA levels were not significantly changed in the uninjured left cortex (data not shown).
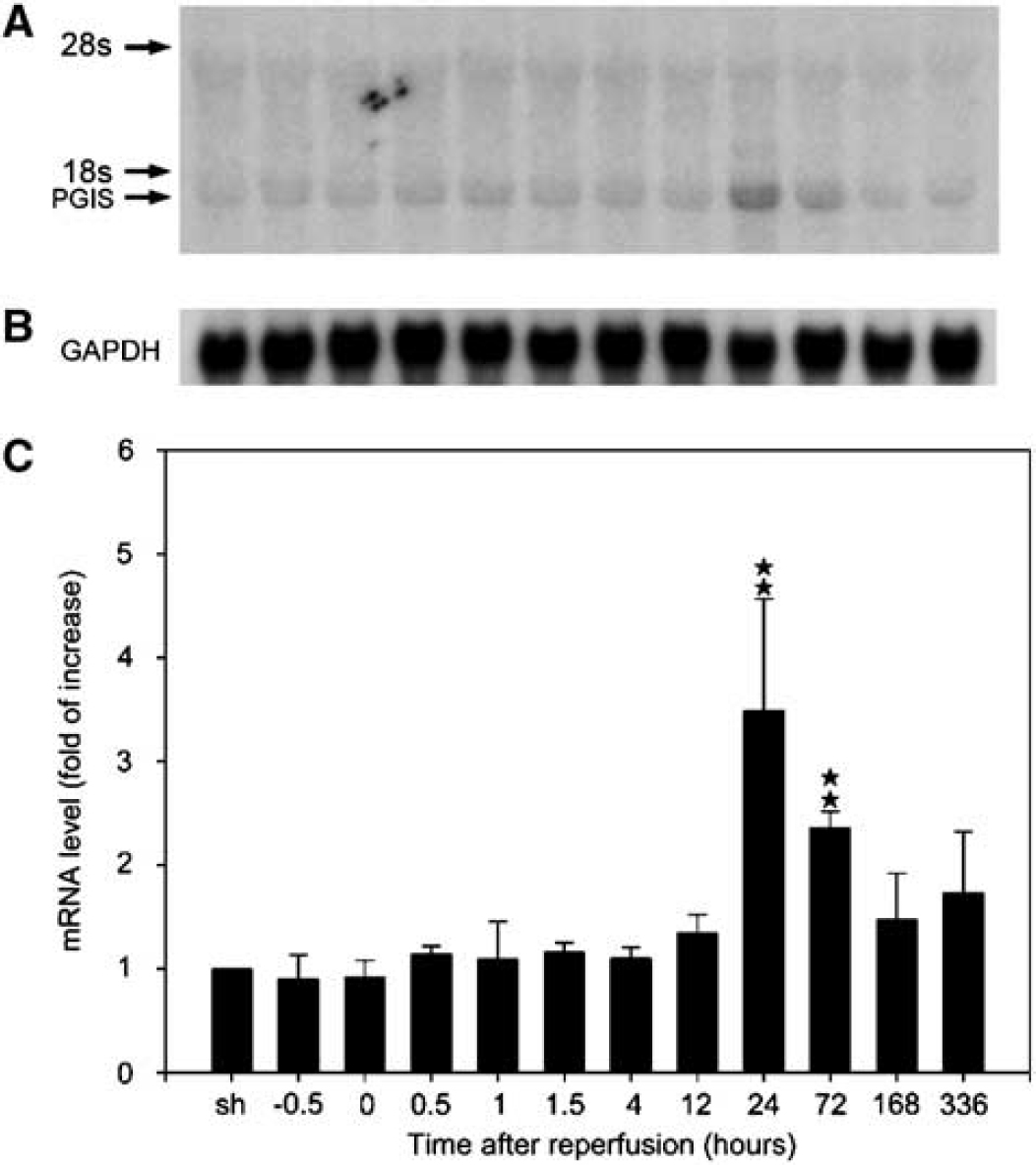
Time-dependent expression of prostacyclin synthase (PGIS) mRNA after brain ischemia–reperfusion. (
In situ hybridization was performed to examine the regional expression of PGIS mRNA. Prostacyclin synthase mRNA signal intensity was very low in the brain of sham-operated controls (Figure 2). At 1-day after ischemia, PGIS mRNA was increased in the penumbra cortical area, particularly the margin demarcating the infarct, the pyriform cortex, the subcortical area (caudate putamen), the hippocampal area, and the leptomeninges of right ischemic hemisphere, all of which became less intense at 72 h. No significant change in the level of PGIS mRNA was noted in the contralateral cortex.
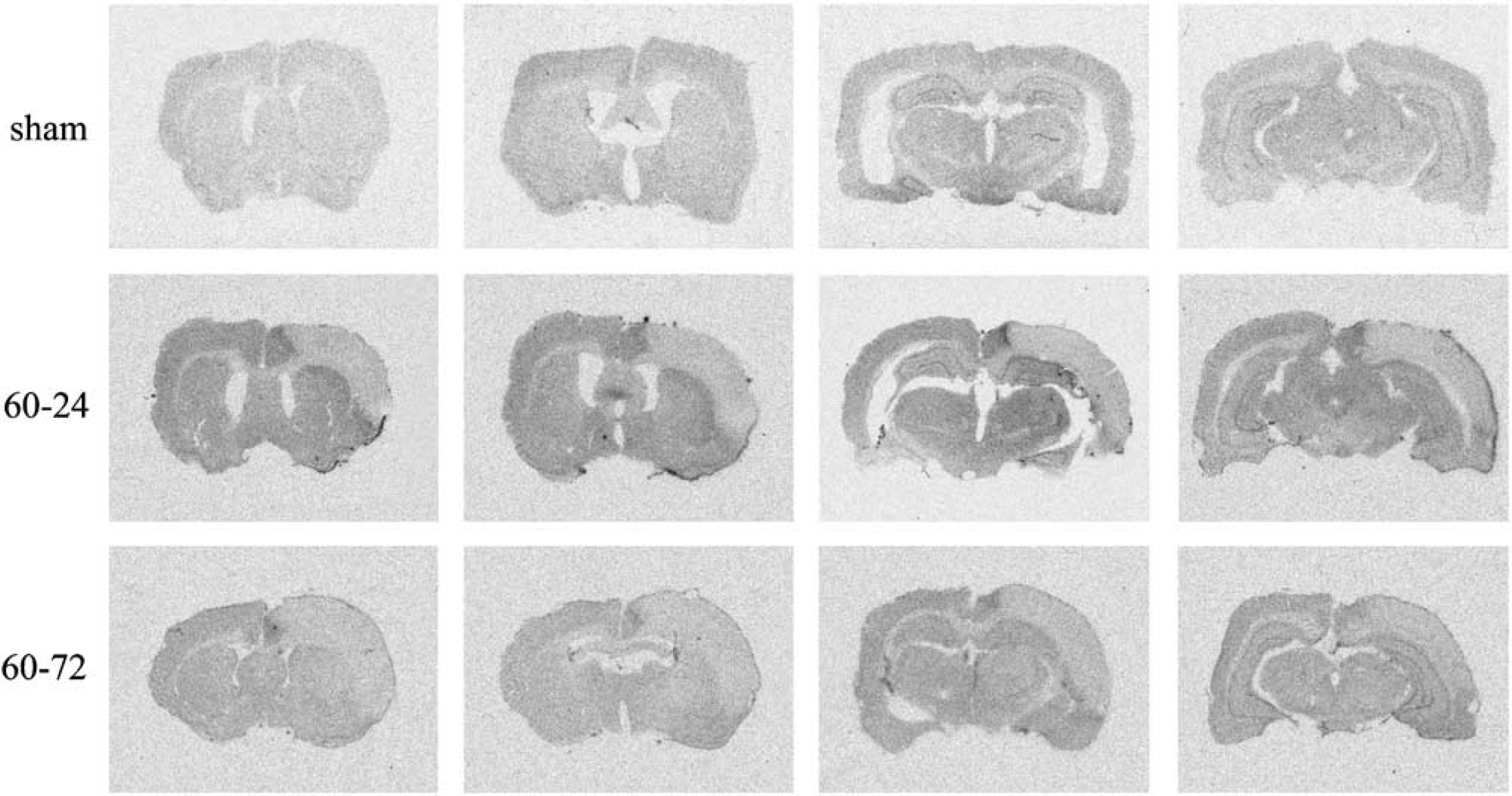
The spatial expression pattern of prostacyclin synthase (PGIS) mRNA in the ischemic brain. Brain slices (25-µm, rostral to caudal) were prepared from rats subjected to 60 mins ischemia followed by 1-day (60 to 24) or 3-day (60 to 72) reperfusion. Brain slices were similarly prepared from sham-operated rats (sham). PGIS mRNA was determined by in situ hybridization as described in Materials and methods. This set of figure is representative of three experiments.
We next examined PGIS proteins in the ischemic brain by Western blot analysis. Very low levels of PGIS proteins were detected at basal and early ischemic time points (Figure 3A). In accord with mRNA levels, PGIS proteins were increased in the ischemic cortex at 24- and 72-h after ischemia (Figure 3A). Quantitative analysis indicates a 23-and 28.5-fold induction at 24- and 72-h reperfusion, respectively, when compared with sham-operated controls (Figure 3C). No significant change in PGIS protein level was noted in the contralateral uninjured cortices (data not shown).
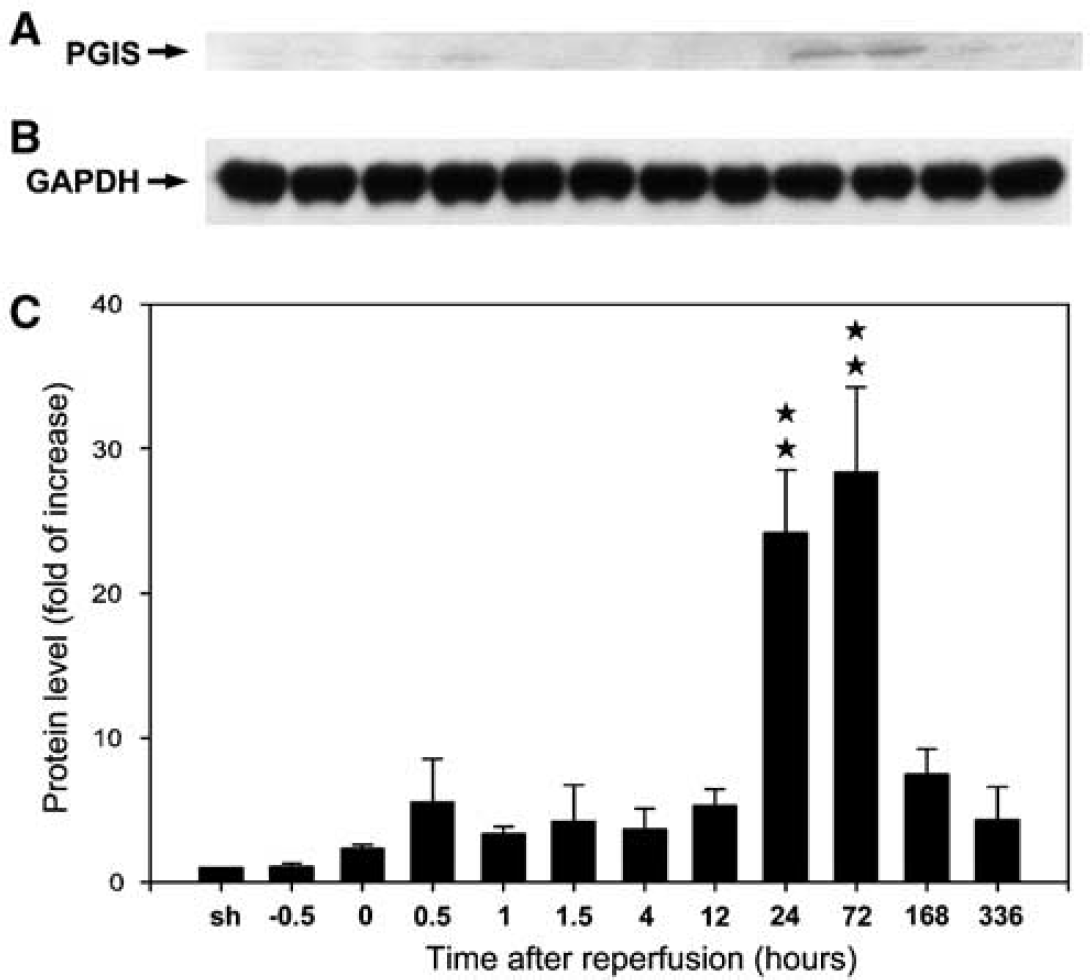
Time-dependent expression of prostacyclin synthase (PGIS) proteins after ischemia–reperfusion. (
The regional expression of PGIS proteins in the ischemic brain was examined by immunohistochemistry. A majority of the PGIS immunoreactivity was detected in the penumbra area of ipsilateral cortex, subcortex (caudate putamen), and the leptomeninges; as well as, albeit to a lesser extent, in the area of ipsilateral hippocampal and pyriform cortex (Figure 4), which corresponds to the spatial expression of PGIS mRNA described above. Furthermore, a substantial amount of capillary-like PGIS staining was also noted within the ischemic cortex (Figure 4E). No significant change in PGIS immunoreactivity was noted in the contralateral uninjured cortex.
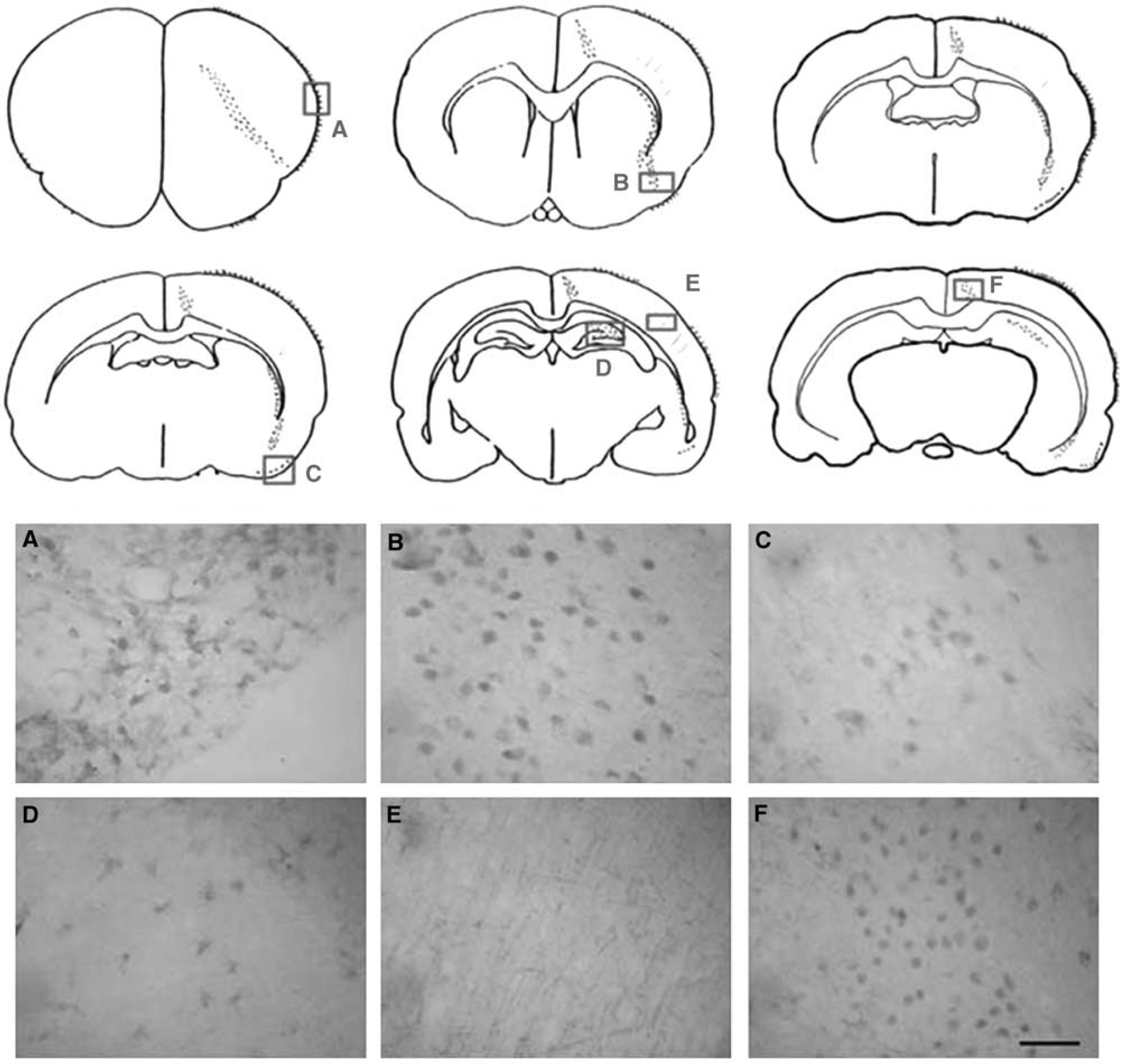
The regional distribution of prostacyclin synthase (PGIS) protein in ischemic rat brains. Coronal sections (25-µm) were prepared from rats subjected to 60-min ischemia followed by reperfusion for 24 h. A schematic drawing of PGIS immunoreactivity in the ischemic brain slices (rostral to caudal) is shown in the upper panel, and the PGIS immunostaining microphotographs at positions
To determine the cellular origin of PGIS immunoreactivity, double immuno-staining was conducted. Prostacyclin synthase immunoreactivity was heavily colocalized with PECAM-1 (an endothelial cell marker) in the leptomeninges (Figure 5A). It was also colocalized with PECAM-1 in large and small vessels, and, to a less extent, in the periinfarct cortical area (data not shown). Colocalization of PGIS with NeuN (a neuron-specific marker) was noted in the pyriform cortex (Figure 5B), peri-infarct cortical area (Figure 5C), subcortical, and CA1 pyramidal cells with diverse appearance. Prostacyclin synthase was also colocalized with OX-42 (a macrophage/microglial marker) in the peri-infarct cortical area (Figure 5D) and leptomeninges, which was less intense when compared with PECAM-1 staining. Furthermore, PGIS was colocalized with GFAP (an astroglial marker) in the hippocampal area (Figure 5E).
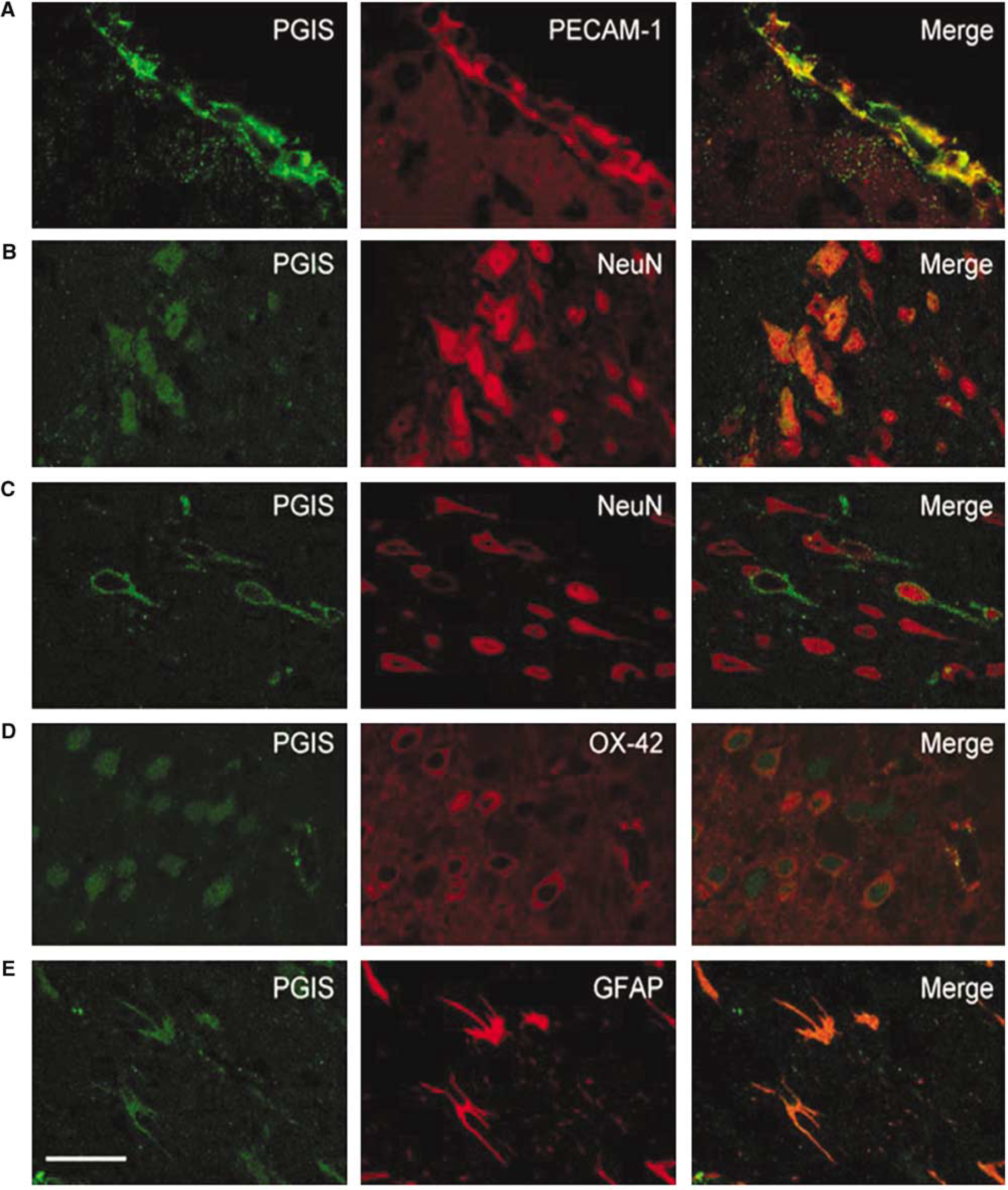
The cellular localization of prostacyclin synthase (PGIS) protein in ischemic brain. Brain slices (25-µm) were prepared from rats subjected to 60 mins ischemia and 24 h reperfusion and double-stained for PGIS and cell-type-specific markers. Representative microphotographs are taken from leptomeninges (
Enhanced PGIS expression in brain tissues after ischemic injury suggests a role of PGIS in ischemic insult. Although bicistronic COX-1/PGIS gene transfer has been shown to protect brain from ischemic injury (Lin et al, 2002a), it is not clear whether PGIS gene transfer alone has a similar protective effect. We, therefore, evaluated the effect of Adv.hPGK-PGIS on infarction. To ascertain the efficiency of adenoviral gene expression via the ventricular administration, we infused Adv.hPGK-PGIS (5 × 107PFU/10 µL) into the right lateral ventricle of normal rats and determined PGIS protein levels 12 to 72 h after administration. Compared with Adv.hPGK control, Adv.hPGK-PGIS augmented PGIS protein levels in a time-dependent manner (Figure 6, lower panel). Maximal augmentation was noted at 48 h after administration. We then investigated the temporal relationship between Adv.hPGK-PGIS treatment and infarct volume. Adv.hPGK-PGIS (5 × 107PFU/10 µL) was infused into the right lateral ventricle at various time windows, and infarct volume was accessed at 24-h after a 60-mins ischemia (Figure 6, upper panel). Significant reduction in infarct volume was noted in rats receiving Adv.hPGK-PGIS 72-h before ischemia. Although a trend of decrease in the infarct volume was noted when Adv.hPGK-PGIS was administered 24-h before, immediately, after or 5 h after ischemia, the reduction was not statistically significant. Paradoxically, the infarct volume increased significantly when Adv.hPGK-PGIS was administered 24-h after ischemia (Figure 6).
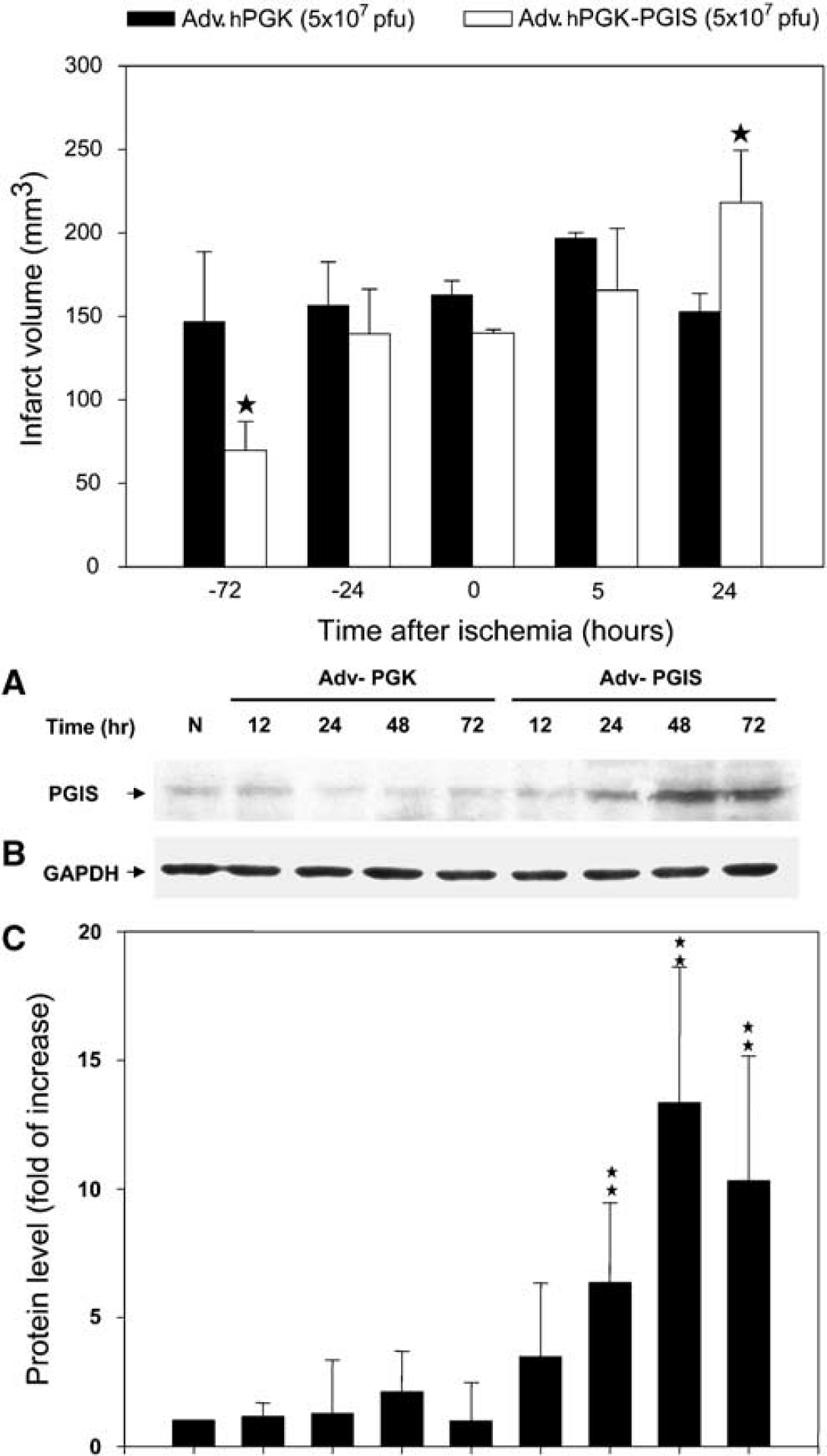
The effect of Adv.hPGK versus Adv.hPGK-PGIS on ischemic brain infarction. Upper panel: Brain infarct volume in rats treated with Adv.hPGK-PGIS or Adv.hPGK at 72 (–72) and 24 (–24) h before; immediately after (0), 5 (5), and 24 (24) h after 60-min ischemia. Data are mean ± s.d. of four rats. *P < 0.05 versus Adv.hPGK controls. Lower panel: Representative Western blots of prostacyclin synthase (PGIS) (
To study the molecular mechanism underlying the beneficial effect of PGIS overexpression, we analyzed eicosanoid levels in the ipsilateral cortex of rats receiving Adv.hPGK-PGIS. The adenoviral vectors were infused intraventricularly into normal rats and eicosanoid levels in the ipsilateral cortex were determined 96 h later. Results show a significant increase in 6-keto-PGF1α and PGE2 (Figure 7A). We next infused adenoviral vectors 72 h before a 60-min MCAO and analyzed eicosanoids in ipsilateral cortex 24 h later (Figure 7B). The total duration of adenoviral exposure in the ischemic brain was also 96 h. Only 6-keto-PGF1α was significantly increased (Figure 7B). PGE2 was higher than control but was not statistically significant (Figure 7B). Because Adv.hPGK-PGIS infused immediately after ischemia failed to reduce infarct volume (Figure 6), we also analyzed the eicosanoids in the ipsilateral cortex 24 h later for comparison (Figure 7B). The results show that besides an equivalent 6-keto-PGF1α elevation, TXB2 level was also elevated (Figure 7B). The levels of other eicosanoids were not altered. This surge of TXB2 level altered the ratio of PGI2 (measured as 6-keto-PGF1α) to TXA2 (measured as TXB2) from approximately 10 in the 72 h preischemic treatment group to approximately 4 in the 0 h postischemic treatment group.
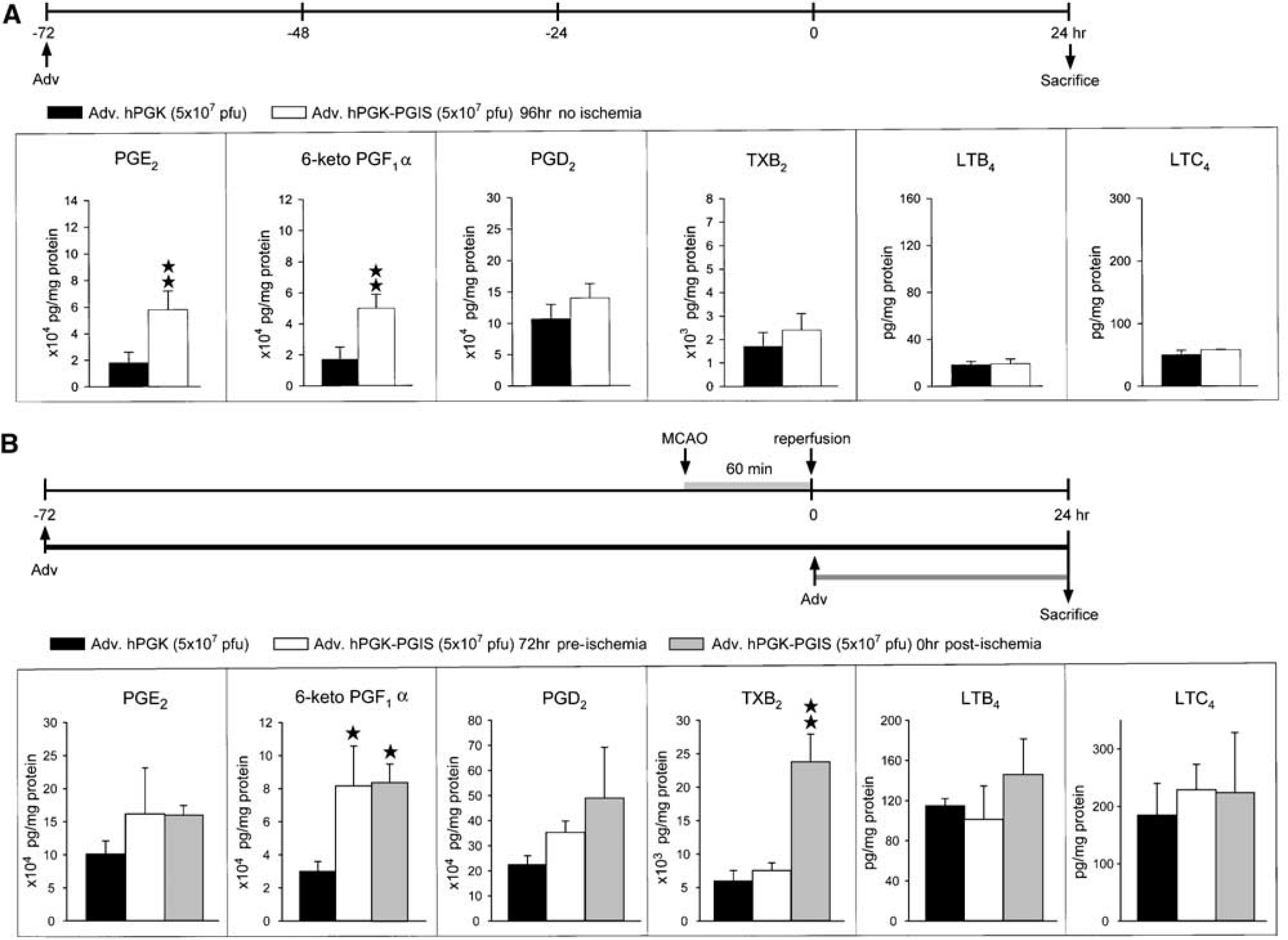
Eicosanoid levels in rat brain transduced with Adv.hPGK-PGIS. (
Discussion
Prostacyclin synthase has been shown to express in the brain of capillaries, blood vessels, choroids plexus, cerebral, and cortical neurons as well as glial cells, such as microglial and oligodendrocytes (Mehl et al, 1999; Siegle et al, 2000). Prostacyclin synthase is generally considered to be constitutively expressed. Its expression has not been reported to be regulated after ischemic insult. In the present study, we report for the first time that brain ischemia–reperfusion injury resulted in a delayed and transient induction of PGIS expression. The RT-PCR-amplified PGIS probe recognized one PGIS mRNA transcript around 2.1 kb, which is in line with the previous report (Tone et al, 1997). Induction of this 2.1 kb transcript was noted at 24- and 72-h after ischemia. Results from in situ hybridization study showed that PGIS mRNA was expressed mainly in the cortical and subcortical regions located in close vicinity of the infarct area, pyriform cortex, hippocampus, and leptomeninges of ischemic right cortex. Western blot analysis and immunohistochemical study confirmed that PGIS proteins were expressed temporally and spatially similar to PGIS mRNAs. The mechanism underlying the late upregulation of PGIS is unclear at the present time. Interestingly, it was reported in this animal model that the postischemic PGI2 levels also peaked at 24-to 72-h of reperfusion (Hsu et al, 1989), which suggests that the late PGIS is functionally active despite the notion that COX-1 and PGIS undergo suicidal inactivation during catalysis (Sanduja et al, 1994; Wang and Chen, 1996) and inactivation of PGIS by peroxynitrite (Zou et al, 1999).
At 24 h after ischemia, a substantial amount of capillary-like distribution of PGIS immunoreactivity was detected within the ischemic MCA cortex. Cellular localization study showed that PGIS immunoreactivities were heavily colocalized with PECAM-1 (endothelial marker)-positive cells at blood vessels, in particular the leptomeninges. It has been shown that PGIS is predominantly expressed in small and large blood vessels throughout the brain (Mehl et al, 1999; Siegle et al, 2000). Prostacyclin synthase-null mice developed ischemic disorders, including arterial sclerosis and hypertrophy of vessel walls by thickening the thoracic aortic media and adventitia (Yokoyama et al, 2002). Mice lacking PGI2 receptor (IP) had a larger ischemic myocardial infarction than wild-type mice (Xiao et al, 2001). Prostacyclin analogs reduced ischemic neuronal damage by increasing blood flow (Cui et al, 1999). In the present study, our results show that a majority of PGIS increment was detected in the endothelial cells after ischemic insult, which confirmed an important role that endothelial cells play in protecting brain tissues from ischemic insult, at least in part via PGI2.
Our results show that PGIS was also colocalized with NeuN (neuronal marker) in the penumbra cortex, hippocampal, and caudate putamen area. Although PGIS has been shown to express in the cortical neuron (Mehl et al, 1999; Siegle et al, 2000), the exact physiologic significance is not clear. Nevertheless, it has been shown that PGI2 and its analogs protect neuronal cells against ischemic insult by inhibiting excessive calcium influx to neurons (Pluta, 1994; Yamashita et al, 1996). Moreover, a subtype of PGI2 receptor (IP) was abundantly expressed in the hippocampus, cerebral cortex, and striatum area, and PGI2 analogs specific for this IP isoform protect neurons against ischemic injury (Cui et al, 1999). Mice lacking IP developed larger ischemic myocardial infarction and less pain sensation than wild-type mice, which were independent of PGI2's effects on platelets (Murata et al, 1997; Xiao et al, 2001). These results suggested a role for neuronal PGIS in modulating cell activity and viability after ischemic insult.
We have reported that COX-1 and bicistronic COX-1/PGIS gene transfer reduced cerebral infarction by augmenting PGI2 (Lin et al, 2002a) and a concomitant reduction in TXA2, LTB4, and LTC4, which are known to contribute to the postischemic cerebral blood flow reduction, brain edema, inflammation, and neuronal damage. It was difficult to tell whether the protective effects conferred by Adv-COX-1 or Adv-COX-1/PGIS is because of an increase in PGI2 level or a reduction in TXB2, LTB4, and LTC4 levels. In the present study, we tested whether PGIS gene transfer exert protective effects by selective augmentation of PGI2 without concurrent alteration in other eicosanoids. The results support this notion. When Adv.hPGK-PGIS was administered 3 days before ischemia, only the PGI2 level was significantly increased in Adv.hPGK-PGIS group. This is in keeping with the previous reports that PGI2 (Matsuda et al, 1997) and stable PGI2 analogs (Cui et al, 1999) attenuated ischemic brain damage in gerbil.
Unlike COX-1 and COX-1/PGIS gene transfer (Lin et al, 2002a), which remains effective in reducing cerebral infarction after ischemia has occurred, we did not detect a significant beneficial effect when Adv.hPGK-PGIS was infused 1-day before or after ischemia. Our results show that both PGI2 and TXA2 levels were increased in brain tissues treated with Adv.hPGK-PGIS immediately after ischemia. It is likely that the concomitant increase in TXA2 neutralizes the PGI2's beneficial effect. The ratio of 6-keto-PGF1α (PGI2) to TXB2 was approximately 10 when Adv.hPGK-PGIS was administered 72-h before ischemia whereas the ratio was reduced to approximately 4 when Adv.hPGK-PGIS was administered immediately after ischemia. This is in line with the notion that PGI2/TXA2 imbalance is deleterious to the circulatory system and lead to vascular diseases such as stroke (de Levai et al, 2004). Nevertheless, the mechanism underlying the increase in TXA2 remains to be studied.
In summary, results of the present study showed that focal cerebral ischemia–reperfusion led to the expression of PGIS at both mRNA and protein levels. Prostacyclin synthase expression occurred in a delayed and transient fashion in the penumbra area including cortical, subcortical, hippocampal, and leptomeninges. Prostacyclin synthase was predominantly expressed in endothelial cells. Enhanced PGIS expression is accompanied by elevated postischemic PGI2 level. Prostacyclin synthase also expressed at neurons in which PGI2 receptors were abundant. Prostacyclin synthase gene transfer increased PGI2 level and reduce cerebral infarction. However, this beneficial effect is time-dependent. Eicosanoid analysis suggests that the time-dependent effect was because of the level of TXA2, especially the ratio between PGI2 and TXA2. Together, these results suggest that PGIS expression after ischemic insult is important in promoting neuronal cell survival.
Footnotes
Acknowledgements
We thank Dr Kenneth K Wu for critically reviewing this manuscript.