
Abstract
Cardiac arrest is often associated with poor neurologic outcome since therapeutic options are limited. We tested the hypothesis that overexpression of CuZn superoxide dismutase (SOD+/–) is neuroprotective in a new murine model of cardiac arrest and cardiopulmonary resuscitation (CPR). Second, we investigated if female and male mice sustain similar injury and if sex-specific outcomes are altered by SOD overexpression. Neuronal injury was quantified 3 days after 8 mins of KCl-induced cardiac arrest by calculating the percentage of ischemic neurons for caudoputamen and hippocampal CA1 region. In rostral caudoputamen, less neuronal cell loss was found for SOD+/– mice (31%+22%) when compared with wild-type (WT) mice (47%+31%, P<0.05). Superoxide dismutase overexpression did not reduce injury in the caudal caudoputamen. No sex-linked protection was evident in either genotype in the caudoputamen. Female WT mice had less CA1 injury than male WT mice (26%+31% versus 54%+30%, P<0.05), whereas no sex difference was found in SOD+/– mice (female: 42%+29%; male: 37%+37%). Comparison of hippocampal injury between genotypes revealed no differences for either males or females. In conclusion, SOD1 overexpression and female sex were associated with significant neuroprotection in this murine cardiac arrest model. However, no additive neuroprotection was observed, and these beneficial effects were restricted to specific brain regions.
Keywords
Introduction
Each year, about 500,000 people suffer a cardiac arrest in the United States, an event which is associated with high mortality and poor neurologic outcome (Eisenberg and Mengert, 2001; Roine et al, 1993). Female sex is associated with a lower incidence of cardiac arrest (Kim et al, 2001; Perers et al, 1999; Wigginton et al, 2002), but men and women have similar mortality rates, neurologic outcome, and quality of life after cardiac arrest (Nichol et al, 1999; Herlitz et al, 2001), although some studies found female sex to be associated with a slight improvement in survival (Herlitz et al, 2001; Vukmir et al, 2003). Despite improvements in resuscitation techniques, survival rates have not changed for decades (Herlitz et al, 2001; Rea et al, 2003). One reason for this disappointing development is the lack of effective treatment options to ameliorate reperfusion injury in the postresuscitation period despite promising results of a variety of agents in animal studies (Jastremski et al, 1989; Roine et al, 1993). However, recent clinical trials showed that induction of mild hypothermia in unresponsive cardiac arrest survivors can improve neurologic outcome and 6-month survival (Hypothermia after Cardiac Arrest Study Group, 2002). This was the first demonstration in humans that the development of brain injury after cardiac arrest can be positively influenced by a postischemic intervention, even with delayed onset of treatment.
Superoxide anion has long been recognized as a key oxidant species in reperfusion injury after cerebral ischemia (Chan et al, 1996; Kontos, 2001; Lewen et al, 2000). Numerous sources of enhanced superoxide production have been identified, including anoxic mitochondria, activated microglia and neutrophils, and membrane-bound oxidases (Lewen et al, 2000; Lipton, 1999). Under physiologic conditions, superoxide anion is processed to hydrogen peroxide by superoxide dismutase (SOD). Three isoforms of SOD have been described, cytosolic CuZn SOD (SOD1), mitochondrial Mn SOD (SOD2), and extracellular SOD. Under pathologic conditions, excessively produced superoxide anions can react directly with cellular components or react with nitric oxide (NO) to form peroxynitrite, a highly reactive oxidant, capable of damaging all major components of a cell, for example, initiation of lipid peroxidation, inhibition of mitochondrial respiratory chain enzymes, oxidative protein modifications, and DNA strand breakage (Szabo, 2003). Superoxide dismutase is the only known component able to compete effectively with NO for superoxide anions and thus prevent formation of peroxynitrite (Beckman et al, 1994). Using either mice or rats overexpressing SOD1 or SOD1-knockout mice, it has been shown that SOD1 exerts neuroprotection in models of traumatic brain injury and transient global or focal cerebral ischemia (Chan et al, 1996, 1998; Igarashi et al, 2001; Kawase et al, 1999; Kondo et al, 1997; Murakami et al, 1997; Sampei et al, 2000; Sugawara et al, 2002; Yang et al, 1994).
We recently developed a new murine cardiac arrest/cardiopulmonary resuscitation (CPR) model (Burne-Taney et al, 2003; Kofler et al, 2004), which more closely reflects the human clinical situation of complete cardiovascular standstill than vessel occlusion models, and aimed to test the hypothesis that SOD1 overexpression is neuroprotective in these experimental conditions of whole body ischemia. The majority of studies exploring the actions of SOD in ischemia were performed in male animals; but in light of emerging evidence that there are sex-specific differences in response to antioxidant treatment (Igarashi et al, 2001; Kavanagh and Kam, 2001; McCullough et al, 2002; Sampei et al, 2000), we also aimed to investigate if female and male mice sustain similar injury, and if any sex-specific outcomes are altered by SOD overexpression.
Methods
This study was conducted in accordance with the National Institutes of Health guidelines for the care and use of animals in research, and protocols were approved by the Animal Care and Use Committee at Johns Hopkins Medical Institutions.
Experimental Groups
Heterozygous hSOD1 transgenic mice (SOD+/–, 25 males, 18 females), which carry the human copper-zinc SOD gene, were compared with age-matched C57Bl/6J wild-type mice (WT; 33 males, 24 females; obtained from Charles River). The SOD+/– mice were produced as previously described (Wong et al, 1995). These animals were originally produced in the C57Bl/6JxHeJ hybrid strain, initially backcrossed to this same hybrid, and then subsequently bred to the C57Bl/6J strain. The level of hSOD1 to endogenous mouse SOD1 activity in brain is 8:1, with a transgene product distribution similar to that of endogenous mouse enzyme (Wong et al, 1995). The genotype of all mice was determined by polymerase chain reaction (PCR) as previously published (Sampei et al, 2000).
Cardiac Arrest Model
Anesthesia was induced with 3% halothane and maintained with 1% to 1.5% halothane in oxygen-enriched air (FiO2 30%) via face mask. Temperature probes were inserted into the left temporalis muscle and into the rectum. Rectal temperature was controlled at near 37°C during surgery with a heating lamp and a heating pad. For drug administration, a PE-10 catheter was inserted into the right internal jugular vein and flushed with heparinized 0.9% saline solution. Another PE-10 catheter was introduced into the right femoral artery, flushed with heparinized 0.9% saline solution, and connected to a pressure transducer system for continuous monitoring of arterial blood pressure (Gould Instruments, Valley View, OH, USA). After completion of surgery, the animals were endotracheally intubated using a 22 G intravenous catheter and connected to a mouse ventilator (Model 687, Harvard Apparatus, Holliston, MA, USA) set to a respiratory rate of 160/min. Tidal volume was adjusted to maintain arterial carbon dioxide tension within the physiologic range (35 to 45 mm Hg).
Animals were then allowed to stabilize for at least 10 mins before baseline temporalis and rectal temperatures and mean arterial blood pressure (MABP) were recorded. Cardiac arrest was induced by injection of 70 μL of cold (4°C) 0.5 mol/L KCl via the jugular catheter, and confirmed by the immediate drop of arterial blood pressure. The endotracheal tube was then disconnected from the ventilator, and anesthesia was ceased. Because preliminary experiments showed that 8 mins of normothermic cardiac arrest resulted in minimal neuropathologic injury, we decided to heat the head of the animal during the cardiac arrest to increase neuronal damage. Temperature was raised by use of a water-filled coil, which was placed around the animal's head and heated to 41°C by running through a water bath (Kofler et al, 2004). Target temporalis temperature was set to 39.5°C to 40°C. Preliminary experiments also showed that body hypothermia greatly improves resuscitability and survival. Therefore, the animals were cooled during ischemia by use of an alcohol patch placed on the front side of the body and a water-filled pad placed underneath, which was chilled by running through an ice-water bath. Body temperature was allowed to decrease until 27°C, then the cooling devices were removed.
Eight minutes after the induction of cardiac arrest, CPR was begun by injection of 0.5 mL prewarmed epinephrine solution (16 μg epinephrine/mL 0.9% saline), chest compressions at a rate of approximately 300/min, and ventilation with 100% oxygen at a respiratory rate of 190/min. As soon as restoration of spontaneous circulation (ROSC) was achieved, defined as a sustained systolic arterial blood pressure of 60 mm Hg (Idris et al, 1996), cardiac massage was ceased. In case of sustained cardiac arrest, additional doses of 0.2 mL epinephrine solution were administered in 1 min intervals. If ROSC could not be achieved within 2.5 mins of CPR, resuscitation efforts were abandoned. Simultaneously with the beginning of CPR, the head temperature control system was switched to cooling and finally turned off when a temporal temperature of 37°C was reached. The body was rewarmed using the heating lamp and pad at a rate of 0.3°C to 0.5°C/min. Twenty minutes after ROSC, both catheters were removed, and the skin wounds were closed. Thirty minutes after ROSC, mechanical ventilation was stopped and the endotracheal tube removed after gentle suctioning. In case of insufficient spontaneous breathing, mechanical ventilation was continued until a respiratory rate of at least 30/min was reached. After extubation, the animal was placed into its homecage for complete recovery.
Histologic Evaluation
Three days after cardiac arrest, animals were deeply anesthetized with intraperitoneal pentobarbital and transcardially perfused with 0.9% saline followed by 10% phosphate-buffered formalin. The brains were carefully removed, postfixed in formalin and embedded in paraffin. Coronal sections 10 μm thick were serially cut with a vibratome and stained with hematoxylin and eosin. With the investigator masked to the genotype of the animal, injury to the CA1 sector of the hippocampus at bregma −1.5 mm, and the rostral and caudal caudoputamen (bregma 0.5 and −1.0 mm, respectively) were evaluated by light microscopy (x 100 objective). Nonviable neurons were considered to have pink eosinophilic cytoplasm and a dark pyknotic nucleus. Viable and nonviable neurons were counted manually, and the percentage of nonviable neurons was calculated for each area of interest. For assessing hippocampal injury, the entire length of the CA1 sector was evaluated. For striatal damage, six microscopic fields were investigated on both levels, respectively, following a distinct pattern. Values from the hemisphere with the worse damage were used for the final analysis.
Western Blot Analysis
Western blot analysis with anti-SOD1 antibody was used to determine if there were regional and sex differences in SOD1 levels of expression. Brains from male and female WT mice (n = 4 per group) were dissected on ice to isolate hippocampus and caudoputamen and frozen in 2-methyl***butane over dry ice. Frozen samples were homogenized in RIPA lysis buffer (50 mmol/L Tris, 150 mmol/L NaCl, 2 mmol/L EDTA, 0.5% Triton X-100, 0.25% sodium-deoxycholate, 1 mmol/L sodium fluoride, 1 mmol/L sodium orthovanadate, 1 mmol/L phenylmethylsulfonylfluoride (PMSF), in the presence of protease inhibitor cocktail (Complete Mini, Roche, Indianapolis, IN, USA)), and centrifuged at 15,000g for 20 mins at 4°C. Protein concentration in the supernatant was measured by BCA protein assay (Pierce, Rockford, IL, USA). Samples of protein (50 μg) were boiled in Laemmli sample buffer (Bio-Rad, Hercules, CA, USA) for 2 mins, loaded on 15% Tris-HCl gels (Bio-Rad, Hercules, CA, USA), separated by electrophoresis, and blotted onto polyvinylidene difluoride (PVDF) membranes (Immobilon transfer membrane, Millipore, Bedford, MA, USA). Blots were blocked with 5% nonfat dry milk in PBST for 1 h at room temperature and then incubated with rabbit anti-Cu/Zn SOD polyclonal antibody (1:750; overnight at 4°C; Stressgen, Victoria, British Columbia, Canada) followed by incubation with goat anti-rabbit IgG horseradish peroxidase conjugate (1:5,000; 1 h at room temperature; Bio-Rad, Hercules, CA, USA). Blots were incubated with enhanced chemiluminescence detection reagents (Amersham Biosciences, Piscataway, NJ, USA) for 1 min and visualized on Kodak Image Station 2000R (Eastman Kodak, Rochester, NY, USA) after exposure for 5 mins. The optical density of the SOD1 bands was measured using Kodak 1D Image Analysis Software (Eastman Kodak, Rochester, NY, USA). Mouse brain tissue extract (Stressgen, Victoria, British Columbia, Canada) was used as a positive control.
Statistical Analysis
All data were expressed as mean+s.d. Physiologic variables and histology were analyzed by one-way ANOVA followed by a Tukey's post hoc test. Injury between rostral and caudal caudoputamen was compared by a paired t-test. The degree of correlation of damage between hemispheres and between areas, and the correlation of injury to duration of CPR was evaluated by calculating Pearson's correlation coefficient. Recovery rate from cardiac arrest and mortality were compared between groups by a χ2-test. Significance was assumed with P<0.05.
Results
Injection of KCl led to immediate systolic cardiac arrest in all mice. All but 2 mice could be successfully resuscitated within the predefined time window of 2.5 mins, which is equivalent to an overall recovery rate from cardiac arrest of 98%. Most mice were resuscitated within 1 min of CPR efforts. Mean CPR time and epinephrine dose were similar in all study groups and are shown in Table 1. The overall 3-day survival rate was 71% with no significant differences between groups. No differences were seen between survivors and nonsurvivors with respect to cardiac arrest-related parameters.
Physiological and cardiac arrest related parameters
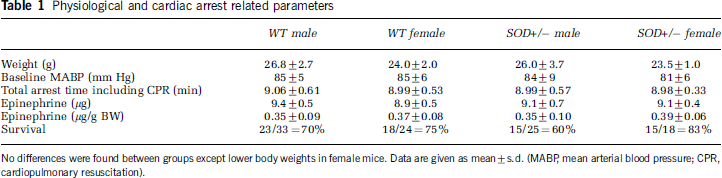
No differences were found between groups except lower body weights in female mice. Data are given as mean±s.d. (MABP, mean arterial blood pressure; CPR, cardiopulmonary resuscitation).
Body weights and baseline MABP values of animals surviving until transcardiac perfusion are given in Table 1. Except for slightly lower body weights in female mice, there were no differences between groups. Arterial blood pressure (Figure 1), temporalis and rectal temperatures were similar in all groups throughout the experiment. Because temporalis temperature was not controlled after ROSC, cooler blood flow from the body to the brain resulted in moderately hypothermic head temperatures during the early reperfusion period.
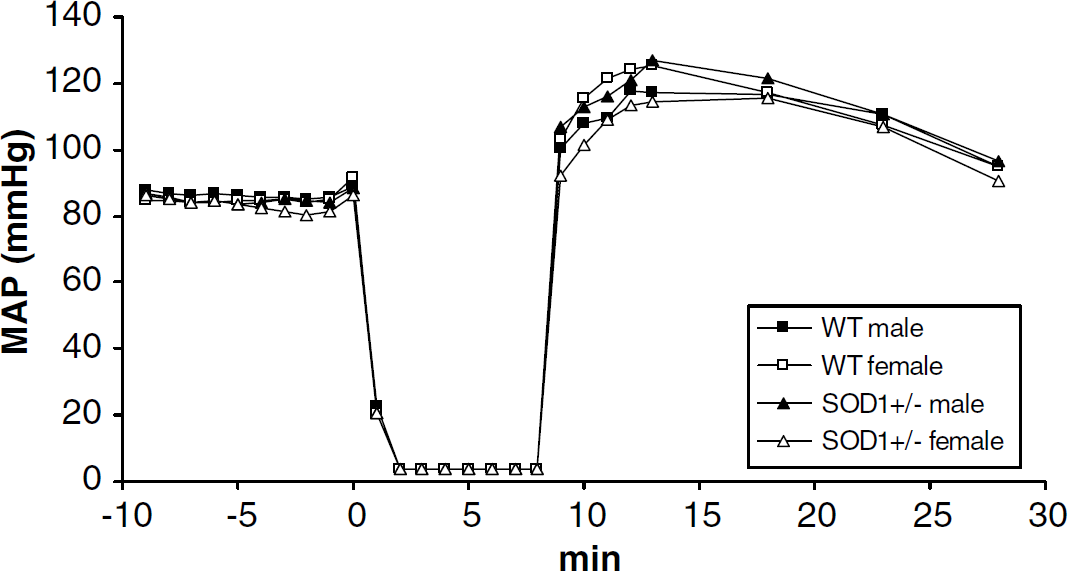
Mean arterial blood pressure (MABP) before, during, and after cardiac arrest. No differences between groups were found at any time point.
All mice regardless of genotype or sex sustained histologic injury. In the rostral caudoputamen, no differences in injury were found between males and females within each genotype. Therefore, both sexes were combined to test for effect of genotype. This analysis revealed that SOD1 overexpression reduced striatal injury (WT: 47%+31%, SOD+/–: 31%+22%, P<0.05; Figures 2 and 3). No differences between groups were found for the caudal caudoputamen.
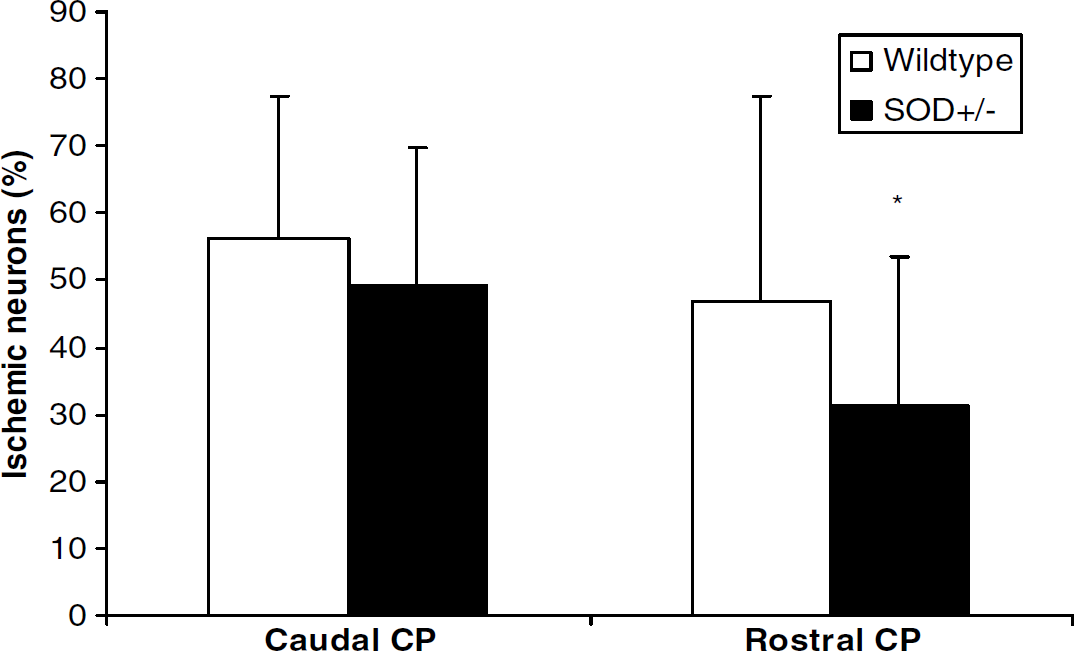
Percentage of damaged neurons after 8 mins of cardiac arrest and 3 days of reperfusion comparing WTand SOD+/– mice. SOD1 overexpression reduced neuronal cell loss in the rostral, but not in the caudal caudoputamen. Data are presented as mean+s.d., *P<0.05 compared with WT.
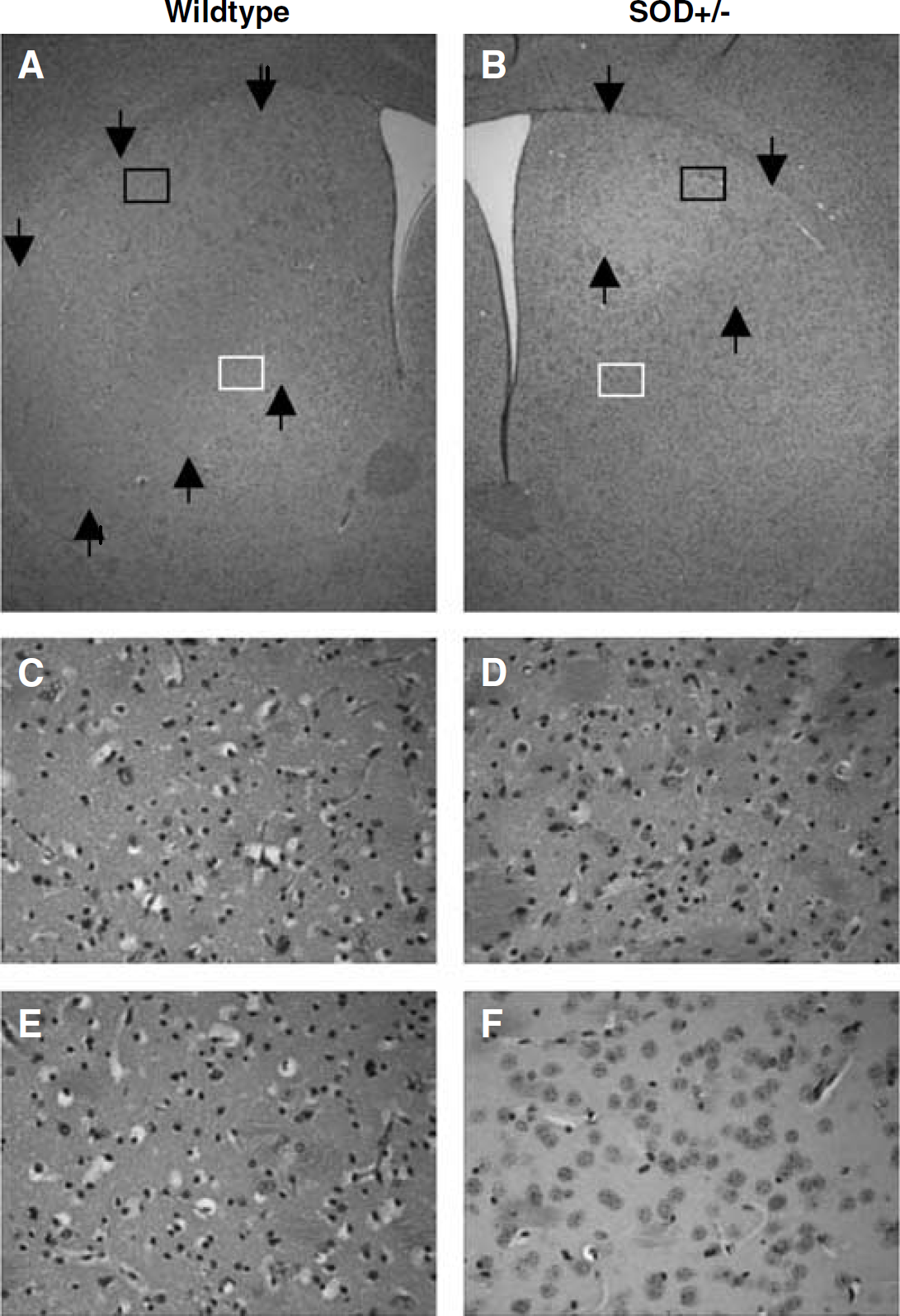
Representative photomicrographs showing ischemic damage in the rostral caudoputamen stained with hematoxylin and eosin at 3 days after cardiac arrest. In the WT mouse (left column), only a small ventral part of the caudoputamen is free of injury, whereas in the SOD+/– mouse (right column) the injury is confined to the dorsolateral area. Injured areas are indicated by arrows. Black boxes in pictures
Neuronal loss appeared in a patchy pattern with islands of resistant cells. No frank infarction was observed in caudoputamen. In general, the dorsolateral and central areas of caudoputamen were most severely damaged. Besides the ventrodorsal and mediolateral gradients, we also found a rostrocaudal gradient, since the percentage of injured cells was higher in the caudal than in the rostral caudoputamen (rostral caudoputamen: 40%+28%, caudal caudoputamen: 53%+20%, P<0.001).
The reduction of injury in SOD+/– mice compared with WT mice was not so much because of a decreased density of ischemic cells, but mainly to a decreased extent of injury (Figure 3). Separate analysis of the distinct counting fields revealed that protection was mainly achieved in the ventromedial subregions of the rostral caudoputamen, whereas in the dorsolateral areas the injury was similar in both genotypes.
Analysis of the CA1 subregion of hippocampus revealed that female WT mice had less injury than male WT mice (male: 54%+30%, female: 26%+31%, P<0.05; Figures 4 and 5). No differences were seen between male and female SOD+/– mice (male: 37%+37%, female: 42%+29%). Because of the sex-specific responses in the WT group, we analyzed the effect of SOD overexpression for each sex separately and found no significant differences between genotypes for either males or females (Figure 5).
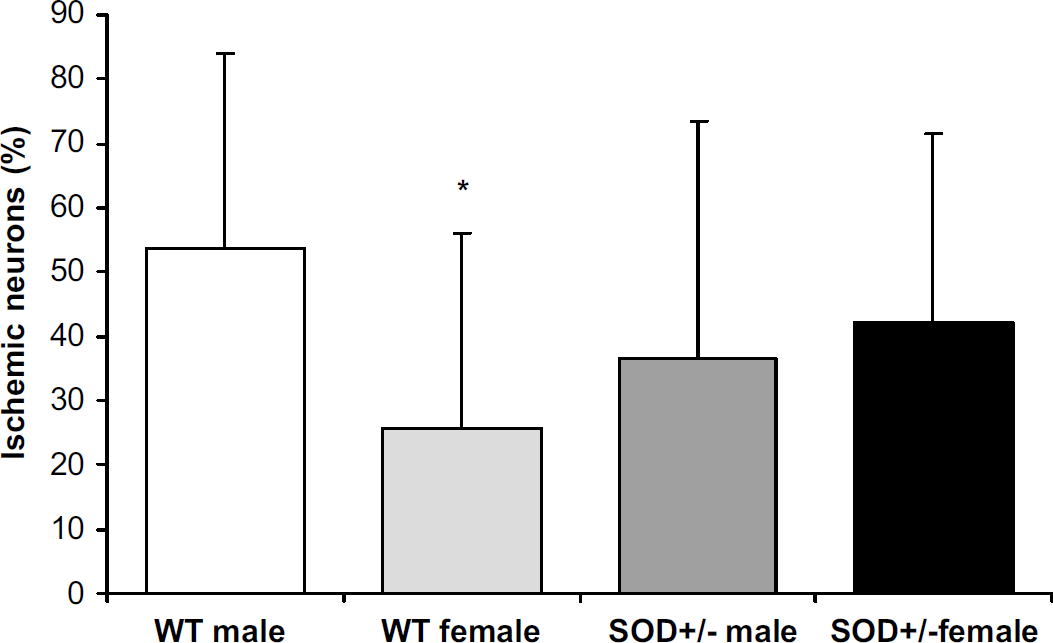
Percentage of damaged CA1 neurons after 8 mins of cardiac arrest and 3 days of reperfusion comparing male and female mice of each genotype. Female WT mice have less injury than male WT mice. Data are presented as mean+s.d., *P<0.05 compared with WT male.
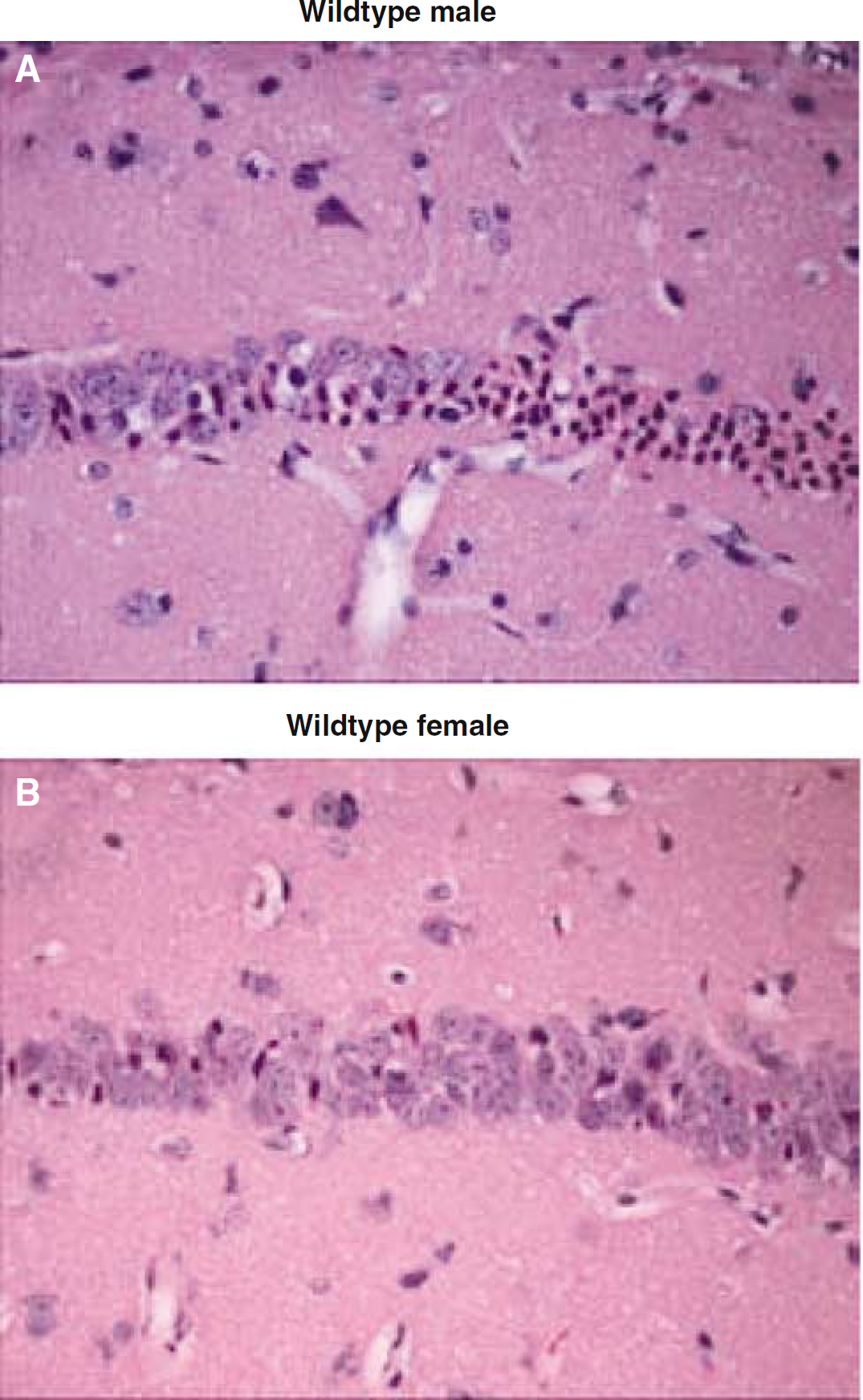
Representative photomicrographs showing ischemic damage in the CA1 subfield of the hippocampus stained with hematoxylin and eosin. At 3 days after cardiac arrest, most of the CA1 neurons in the male WT brain (
To further characterize the model, we compared degree of injury between left and right hemispheres and found good correlation for all areas (CA1: r = 0.829, P<0.001; rostral caudoputamen: r = 0.928, P<0.001; caudal caudoputamen: r = 0.756, P<0.001). We also found a good correlation between the rostral and caudal parts of the caudoputamen (r = 0.628, P<0.001). There was a weaker, but still significant correlation between CA1 and the two areas of the caudoputamen (rostral caudoputamen: r = 0.359, P<0.01; caudal caudoputamen: r = 0.267, P<0.05). Overall, mice resuscitated within 1 min of CPR efforts had significantly less injury in caudoputamen than mice with a low-flow period >1 min (rostral caudoputamen: 33%+29% versus 53%+22%, P<0.01; caudal caudoputamen: 49%+22% versus 64%+19%, P<0.01). For hippocampus, this trend was not significant (<1 min: 38%+31%, >1 min: 49%+36%).
Western blot analysis revealed that the level of expression of SOD1 was similar in hippocampus and caudoputamen in WT mice, as illustrated in Figure 6. No differences in SOD1 protein level were found for either brain region between males and females.
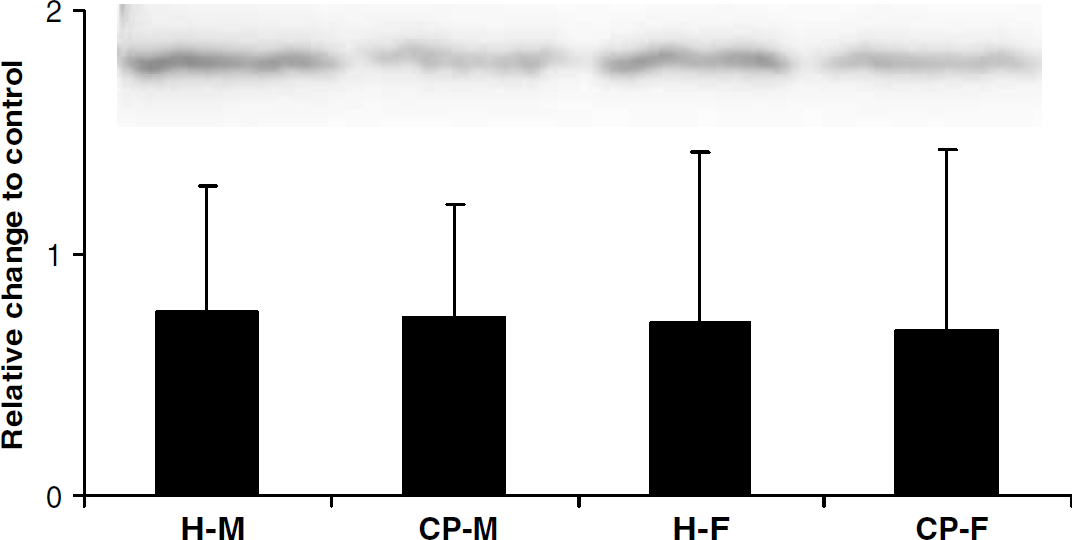
Expression of SOD1 protein in brains from naïve male (M) and female (F) mice, dissected into hippocampus (H) and caudoputamen (CP). The graph indicates change in optical density compared with positive control (mouse brain extract). No differences in SOD-1 protein expression were seen between brain regions or sexes. Values are mean+s.d. of four experiments, respectively. The inset shows a representative Western blot.
Discussion
In the present study, we found that SOD1 overexpression reduced neuronal cell loss in caudoputamen after global cerebral ischemia because of cardiac arrest/CPR. No neuroprotection was observed in hippocampus by SOD1 overexpression. Secondly, female WT mice exhibited less injury in CA1 when compared with WT males. This sex difference was completely lost in SOD+/– mice. No sex-linked protection was evident in either genotype in caudoputamen.
Numerous studies have shown that female sex and administration of estrogen are associated with neuroprotection after focal and global cerebral ischemia (Alkayed et al, 1998; He et al, 2002; Hurn and Macrae, 2000; Jover et al, 2002), although one study reported an opposite effect of estrogen (Harukuni et al, 2001). In our WT mice, we observed a sex-linked neuroprotective effect in hippocampus, but not in caudoputamen. This regional difference might be explained by the fact that expression and density of estrogen receptors (ER) vary throughout the brain. While hippocampal CA neurons express ER-α and ER-β, contradictory results are reported regarding the presence of ERs in caudoputamen (Küppers and Beyer, 1999; Mufson et al, 1999; Shugrue et al, 1997). Further studies using ER-subtype knockout mice are warranted to evaluate the contribution of nuclear ERs to region-specific neuroprotection. In vessel occlusion models of focal and global ischemia, decreased injury has also been described in caudoputamen, which was associated with an increase in intraischemic cerebral blood flow in female or estrogen-treated animals (He et al, 2002; Hurn et al, 1995) because of vasodilatory effects of estrogen (Hurn and Macrae, 2000). But in true cardiac arrest with complete circulatory standstill as in our study, this blood flow promoting effect during ischemia is lost. We cannot, however, rule out estrogen effects on blood flow during reperfusion.
Interestingly, while hippocampal neurons in female WT mice were spared compared with WT males, no such effect was observed in SOD+/– mice. A similar loss of sex-specific protection in SOD+/– mice was detected in a study of traumatic brain injury (Igarashi et al, 2001). After focal cerebral ischemia, male mice deficient in neuronal nitric oxide synthase showed neuroprotection compared with WT males, whereas this effect was lost or reversed in female knockout mice (McCullough et al, 2002; Sampei et al, 2000). One possible explanation for the different sex-specific responses to antioxidant treatment might be that estrogen interferes with the free radical cascade at several points. Estrogen can act as a free radical scavenger, and it induces expression of Bcl-2, which has been shown to block cytochrome c release from the mitochondria and decrease the formation of superoxide anions (Cai and Jones, 1998; Kane et al, 1993; Longo et al, 1997). In focal stroke, overexpression of Bcl-2 simulated the protection against ischemic injury conferred by endogenous female sex steroids (Alkayed et al, 2001). Thus, because females already had benefit of the antioxidant effects of endogenous estrogen, it is likely that overexpression of SOD1 did not provide additional protection.
Using either mice or rats overexpressing SOD1 or SOD1-knockout mice, it has been shown that SOD1 reduces brain injury after transient global ischemia (Chan et al, 1998; Kawase et al, 1999; Murakami et al, 1997). While most studies focused on hippocampal injury, one study using male transgenic rats, found neuroprotection in caudoputamen similar to the results of our study (Chan et al, 1998). We evaluated injury at two different rostrocaudal levels of caudoputamen and found that SOD1 overexpression decreased mainly the extent of damaged area, but not so much the density of ischemic neurons. The dorsolateral crescent of striatum has been shown in several studies of global ischemia to be the most vulnerable area of this brain structure (Larsson et al, 2001; Pulsinelli et al, 1982; Zoli et al, 1997). Besides a ventrodorsal and mediolateral gradient, there is also a rostrocaudal gradient towards increased injury (Zoli et al, 1997), which was confirmed by the results of our study, since we found more injury at the caudal level. These regional differences in vulnerability have been attributed to several anatomic and neurochemical features such as regional differences in vascular supply, distribution of glutamergic and dopaminergic inputs and in the varying occurrence of the metabolism/flow uncoupling phenomenon (Zoli et al, 1997). As a consequence of these mechanisms or as an independent factor, the regionally different production of free radicals is also involved in this phenomenon, since, as we show in this study, overexpression of SOD1 can shift this gradient of vulnerability and can diminish the injured area.
For the CA1 section of hippocampus, SOD1 overexpression was also reported to be neuroprotective, although the results were less consistent and dependent on the length of ischemic period and time point of evaluation. In transgenic rats, sustained CA1 protection was observed (Chan et al, 1998). However, in another study in mice it was shown that after a severe ischemic insult, SOD1 overexpression only delayed the maturation of injury, since the protection observed after 1 day was lost after 3 days (Murakami et al, 1997). This might explain why we did not observe significant hippocampal protection.
To evaluate if sex-differences in SOD1 content contribute to hippocampal neuroprotection as observed in our female WT mice, we performed Western blot analysis of caudoputamen and hippocampal tissue isolated from WT mouse brains. We found no differences in levels of SOD1 protein expression between males and females for either brain region. Similarly, SOD1 activity in cytosolic fractions of whole brain homogenates was not affected by gonadectomy or sex hormone therapy (Pajović et al, 1993, 1996). Thus, it appears that sex-associated differences in ischemic cerebral susceptibility are not mediated by different basal expression levels of SOD1.
Because SOD1 overexpression in our transgenic mice was not restricted to neural tissue and, secondly, in cardiac arrest the whole body is affected by the ischemic event, differences in systemic free radical production or scavenging might have influenced our results. It has been shown that SOD1 overexpression also protects the heart against ischemia–reperfusion injury (Wang et al, 1998). However, in preliminary experiments we have not seen any histologic evidence of myocardial cell damage. We did not find any differences between genotypes in blood pressure, recovery from cardiac arrest and mortality, either. It is therefore not likely that any differences in the restoration of cardiac function might have significantly contributed to the improved neurologic outcome in the transgenic mice.
In conclusion, SOD1 overexpression and female sex are associated with significant neuroprotection in this murine cardiac arrest model of global cerebral ischemia. However, these beneficial effects were restricted to specific brain regions. Secondly, no additive neuroprotection was observed but, on the contrary, female sex-associated neuroprotection as seen in WT mice in hippocampus was lost in SOD1 overexpressors. In light of the emerging evidence that there are sex-specific differences in response to antioxidant treatment, future experimental studies should include animals of both sexes. Likewise, in human clinical trials, data should be analyzed separately for males and females, so that any possible sex-associated differences in response to antioxidant therapy are not overlooked.