
Abstract
Cerebral edema is a major contributor to morbidity associated with traumatic brain injury (TBI). The methods involved in most rodent models of TBI, including head fixation, opening of the skull, and prolonged anesthesia, likely alter TBI development and reduce secondary injury. We report the development of a closed-skull model of murine TBI, which minimizes time of anesthesia, allows the monitoring of intracranial pressure (ICP), and can be modulated to produce mild and moderate grade TBI. In this model, we characterized changes in aquaporin-4 (AQP4) expression and localization after mild and moderate TBI. We found that global AQP4 expression after TBI was generally increased; however, analysis of AQP4 localization revealed that the most prominent effect of TBI on AQP4 was the loss of polarized localization at endfoot processes of reactive astrocytes. This AQP4 dysregulation peaked at 7 days after injury and was largely indistinguishable between mild and moderate grade TBI for the first 2 weeks after injury. Within the same model, blood–brain barrieranalysis of variance permeability, cerebral edema, and ICP largely normalized within 7 days after moderate TBI. These findings suggest that changes in AQP4 expression and localization may not contribute to cerebral edema formation, but rather may represent a compensatory mechanism to facilitate its resolution.
INTRODUCTION
Experimental models of traumatic brain injury (TBI) have significantly advanced our understanding of pathophysiology leading to secondary brain injury1–3 and have led to the development of many potential neuroprotective agents. However, few of these neuroprotective strategies have been translated into improved clinical care for patients with acute head trauma and other brain injuries.4–6 Several factors may contribute to the limited success of clinical translation, but one key factor is likely the lack of correspondence between current experimental models and the conditions experienced in real-life accidents. Most patients with TBI sustain their injuries due to blunt, non-piercing head trauma caused by traffic accidents and falls. Penetrating or open head injuries were observed in only 0.8 to 3% of patients.7,8 Unfortunately, the most widely used rodent models of TBI require fixation of the head in a stereotaxic frame, and most variations of controlled cortical impact and fluid percussion injury models require preparation of a cranial window, or are associated with fracturing of the skull. Not only is this different from the typical patient with TBI, but it also prevents the monitoring of intracranial pressure (ICP). Intracranial pressure monitoring is widely used in clinical practice to intercept fatal brain swelling, but cannot be recorded if the skull is already open.
Since first being implicated in the genesis of cerebral edema after ischemic stroke, 9 the astrocytic water channel aquaporin-4 (AQP4) has been the subject of much interest within the TBI field, as its role in post-traumatic edema formation and secondary injury progression has been explored. Under physiologic conditions, AQP4 is expressed primarily on perivascular endfoot processes and the membranes of the glia limitans.10,11 Although the physiologic role of these astroglial water channels remains uncertain, it is now well established that AQP4 plays a key role both in the movement of water into swelling astrocytes,9,12 as in cytotoxic edema, as well as in the resolution of vasogenic edema 13 after ischemic, inflammatory, and other forms of injury. To date, posttraumatic changes in AQP4 expression have not been evaluated within a closed-skull TBI model, making it difficult to accurately assess its role in the formation and resolution of cerebral edema.
In the present study, we first report the development of an experimental murine model that we term ‘Hit & Run’ TBI. This model produces consistent contusion of the temporal lobe without exposing the mice to prolonged anesthesia and without opening or fixing the skull. The injury can be modulated to generate both mild and moderate grades of TBI. Within this model, we detail for the first time the dynamics of AQP4 dysregulation after both mild and moderate closed-skull injury. We further detail complex changes in post-traumatic AQP4 expression and localization in reactive astrocytes that may make important contributions to edema formation and resolution after traumatic injury.
MATERIALS AND METHODS
Animals
This study was carried out in accordance with ARRIVE guidelines. Male C57Bl/6 mice, aged 10–12 weeks (The Jackson Laboratory, Bar Harbor, ME, USA), were used in all experiments. The mice were housed under standard laboratory conditions. All experiments were approved by the University Committee on Animal Resources of the University of Rochester and carried out according to guidelines from the National Institutes of Health.
Hit & Run Device and Injury Model
The device used for ‘Hit & Run’ TBI was modified from the commercially available controlled cortical impact device, also referred to as the Pneumatic Powered Controlled Cortical Impact Device (PPCCID, Pittsburgh Precision Instruments, Pittsburgh, PA, USA). We modified the controlled cortical impact device for Hit & Run trials by rotating the mounting 90° such that the metal rod is positioned horizontally. A polished stainless steel tip, which strikes the mouse's head during the controlled cortical impact, was fitted to the end of the impactor slider. The impacting rod (diameter of 3 mm) can be adjusted to several preset parameters, most importantly including velocity and travel distance (which determines the depth of injury).
In this study, we used a strike depth of 10 mm, and 0.1 seconds of contact time for all injury groups. The velocity was altered as necessary for different severities of injury: 4.8 m/s for mild and 5.2 m/s for moderate TBI. The mouse was anesthetized by 2.5% isofluorane for 2 minutes, then it was hung head up vertically from its incisor teeth by a mounted metal ring after it proved unresponsive to painful stimulation. The impactor was positioned normal to the skull at the loading point halfway between the ipsilateral eye and the external ear canal. Following impact, the mechanism released the injured animal onto a soft pad placed below it. After anesthesia induction, it takes approximately 30 seconds in total to suspend, injure, and release the animal onto the soft pad. The sham injury (control group) mice were anesthetized, hung from the string, and allowed to fall and recover. Arousal time was calculated as the time from cortical impact to spontaneous locomotor activity on the underlying pad. The methodology for evaluating infarct volume, cerebral edema, and ICP is detailed in the Supplementary Methods available on the
Animals were assessed by a gross neuroscore of 3 hours, 24 hours at 3, 7, 14, and 28 days after TBI. Animals underwent rotarod test, open field test, and novel object recognition test 2 days before TBI, then weekly following after TBI for 4 weeks. Barnes maze test was performed one time 28 days after injury. Detailed methodologies for these tests are available in the Supplementary Methods on the
Evaluation of AQP4 Expression and Localization
For a detailed description of the immunofluorescence labeling protocol, see the Supplementary Methods section available on the
For analysis of cortical myelination, 12 ipsilateral and 12 mirror-image contralateral regions were assessed from each injury grade, including sham controls. The × 40 confocal images were acquired as above. Analysis was conducted by individuals masked to the experimental group with ImageJ software as follows: color channels were separated, and the myelin basic protein (MBP) fluorescence channel was background subtracted and uniformly thresholded. To define the extent of myelination, the area of MBP+ labeling was measured and expressed as a percentage of the total image area. To account for differences in labeling, MBP labeling was normalized to values from mirror-image contralateral regions. For SMI-34 labeling, immunolabeling was assessed in regions of reactive gliosis both ipsilateral and contralateral to the TBI lesion by epifluorescence microscopy 14 days after TBI. Representative images were generated from peri-lesional cortical regions at × 40 magnification.
Statistical Analysis
All values are expressed as mean ± s.e.m. Data were statistically evaluated and plotted in the Prism software (Graphpad, La Jolla, CA, USA). Comparisons between injury groups were made by one-way analysis of variance (ANOVA) with Tukey's
RESULTS
Optimization of the Hit & Run Traumatic Brain Injury Model
Hit & run injury was characterized at two different severities: ‘mild’ and ‘moderate’ injuries utilized cortical impactor velocities of 4.8 and 5.2 m/s, respectively, while the impact depth and contact times were held constant. In pilot experiments, impactor velocities were titrated to generate a mild lesion characterized histologically by diffuse cortical reactive astrogliosis, but without frank tissue disruption or cavitation that occurred in the moderate grade injury. After isofluorane anesthesia induction, we found that the injury sequence could be completed rapidly (Figure 1A; mean 19.9 ± 1.9 seconds; range 17 to 24 seconds,
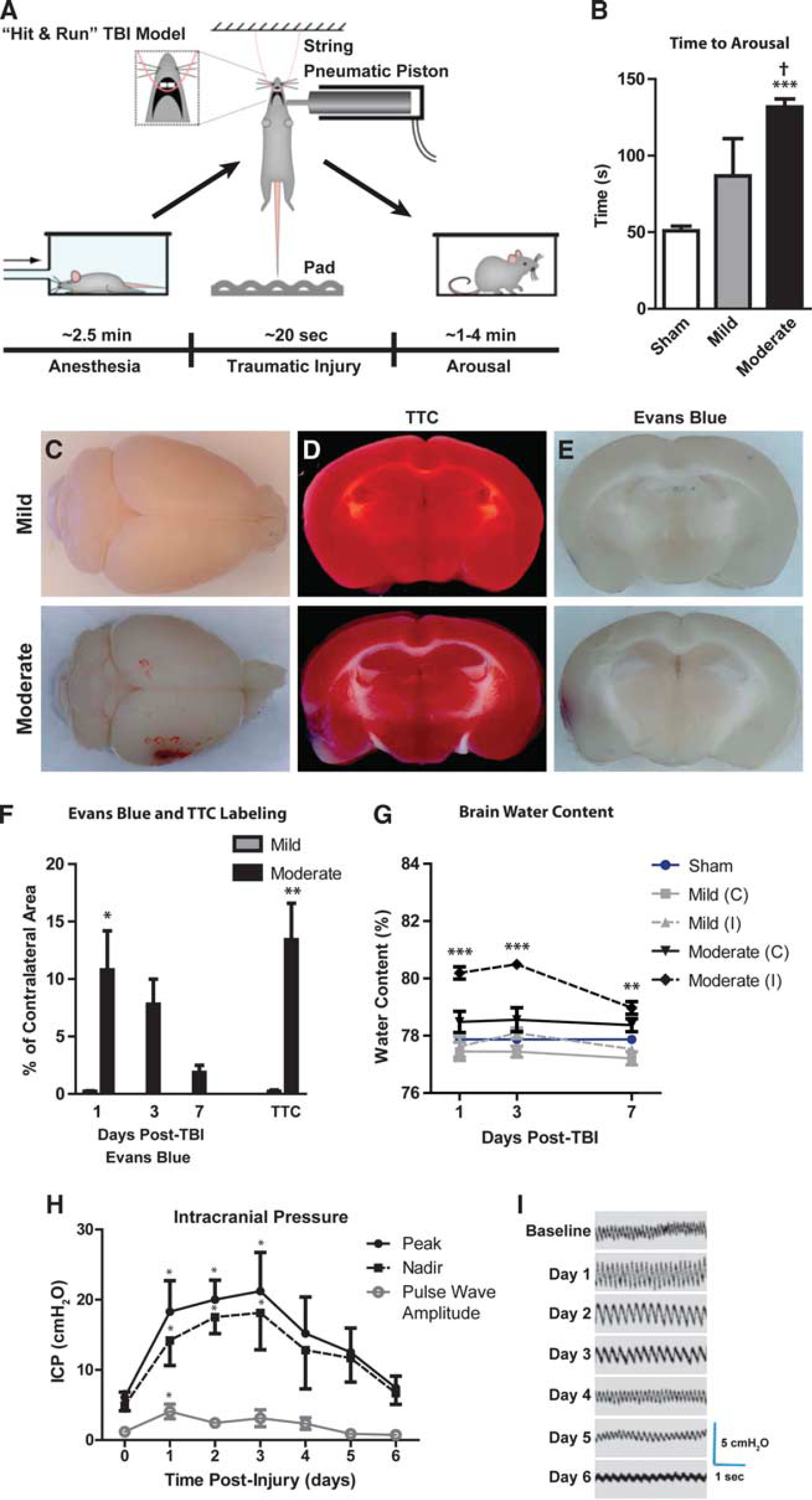
The ‘Hit & Run’ model of murine traumatic brain injury (TBI). (
Mild or moderate Hit & Run injury never resulted in acute death, while delayed death was minimal (one mild and one moderate TBI mouse out of a total of 366 mice). Depressed skull fractures were not observed in mild or moderate TBI animals. Ipsilateral, subdural and/or subarachnoid hemorrhage, and contusion were evident in moderate animals (Figure 1C) localized to the impact site. Twenty-four hours after TBI, the presence of infarction was assessed by triphenyle tetrazolium staining (Figures 1D and 1F). Mild TBI mice did not exhibit appreciable infarction, while moderate TBI animals exhibited small infarcts (~15% of the total slice area; ∗∗
Blood-brain barrier disruption was evaluated after TBI by analysis of Evans Blue extravasation (Figure 1E–F). No appreciable blood–brain barrier (BBB) disruption was evident 24 hours after mild TBI; however, local BBB disruption was evident after moderate TBI that resolved over the first 7 days post-injury (∗
Behavioral Assessments
Mice undergoing Hit & Run TBI were evaluated for cognitive and motor deficits beginning immediately after injury out to 28 days after injury. A gross neuroscore was used to evaluate changes in the animals’ overall physiologic state, motor function, and alertness (Figure 2A). Control animals exhibited no deficits (score of 0) at any time point. Mild TBI animals exhibited neuroscore deficits at 3 and 24 hours after TBI, resolving before 3 days post injury. Moderate TBI animals were significantly impaired compared with mild TBI animals, but global neuroscore in these animals normalized within 7 days post injury (∗
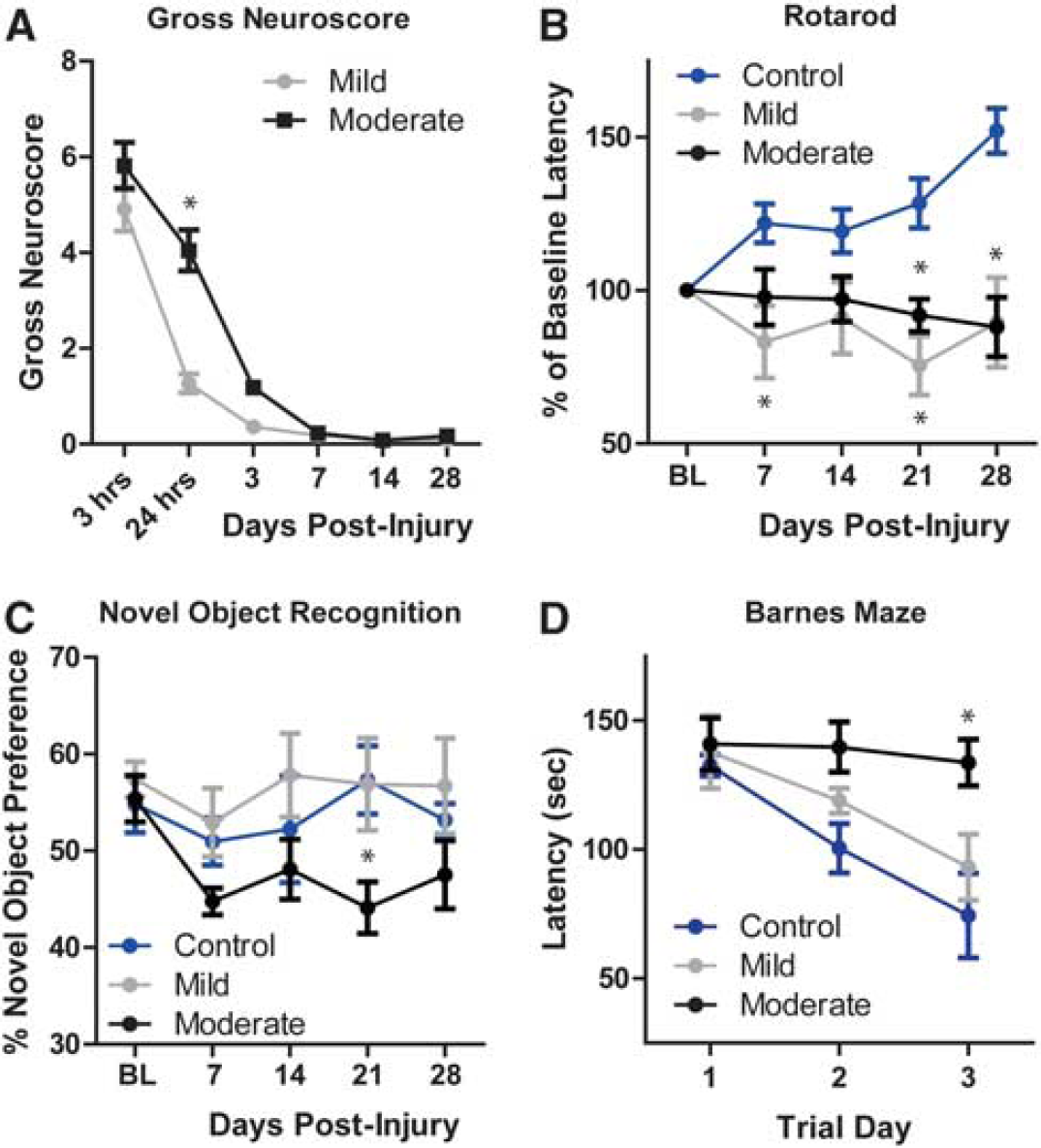
Long-term behavioral deficits in mice undergoing traumatic brain injury (TBI). Behavioral deficits were evaluated in control (sham), mild, and moderate TBI mice for 28 days post injury. (
The novel object recognition test was used to monitor cognitive function each week after TBI (Figure 2C). Control animals exhibited a stable novel object preference in all 5 weeks of study. Mild TBI animals did not exhibit any deficit in the novel object recognition task at any time point; however, moderate TBI animals demonstrated a significant and persistent decline in test performance (∗
Development of Post-Traumatic Reactive Gliosis
We next characterized the development of reactive astrogliosis over the first 28 days after mild and moderate TBI (analysis of 3–6 animals per group, 4 slices per animal). In mild TBI animals 3 days post injury, diffuse GFAP (reactive astrocytes) and CD68 (reactive microglia) labeling were evident local to the impact site, penetrating the depth of the cortex, subcortical white matter, and involving the striatum and hippocampus (Figures 3A and 3C). Cortical reactive astrogliosis and microgliosis remained elevated at 7 days post injury, then resolved over the following 3 weeks (Figures 3B, 3D–3F, and 3K; Supplementary Figure S1A). After mild injury, striatal reactive gliosis followed a similar pattern, peaking 7 days post TBI and resolved through the following 3 weeks (Figure 3L; Supplementary Figure S1B). In contrast, reactive astrogliosis in the hippocampus appeared to persist for up to 1 month after mild injury (Figures 3M–3O). No defined glial scar was apparent in mild TBI animals at any time point.
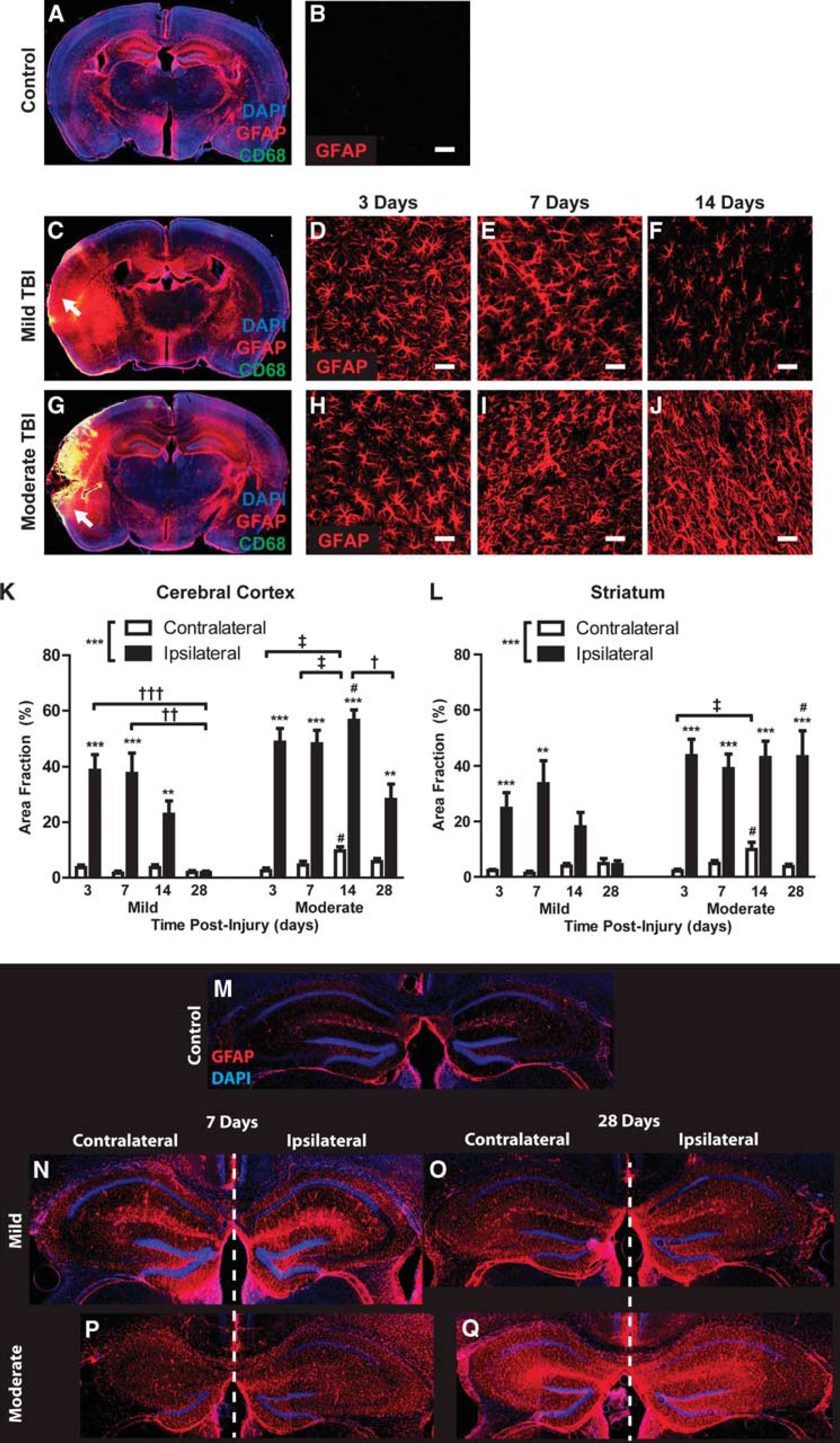
Differing patterns of reactive astrogliosis after mild and moderate traumatic brain injury (TBI). Reactive astrogliosis (glial fibrillary acidic protein (GFAP), red) and microgliosis (CD68, green) were evaluated after mild and moderate TBI. (
In contrast to mild TBI, cortical disruption was apparent in moderate TBI animals 3 days post injury (Figure 3G) that evolved over the first week to form a well-defined glial scar surrounding the lesion cavity, often extending through the subcortical white matter into the striatum. The disrupted cortex at 3 days was continuous with a wide surrounding field of reactive gliosis that encompassed the cortex, subcortical white matter, hippocampus, and striatum. Cortical and striatal reactive gliosis after moderate TBI reached a peak within 3–7 days post injury. Both the extent and persistence of reactive gliosis were significantly greater in moderate TBI compared with mild TBI (Figures 3D–3F and 3H–3J); however, at 7 days post injury, peak reactive astrogliosis did not differ significantly between the mild and moderate conditions (Figures 3K and 3L, #
Demyelination and Axonal Degeneration After TBI
Changes in regional brain volume were measured at 3, 7, 14, and 28 days after mild and moderate TBI. After mild TBI, no measureable changes in cortical or subcortical volume were evident at any time point (Figures 4A and 4B). After moderate TBI, a delayed loss of cortical tissue was observed beginning 7 days after injury (Figure 4A; ∗
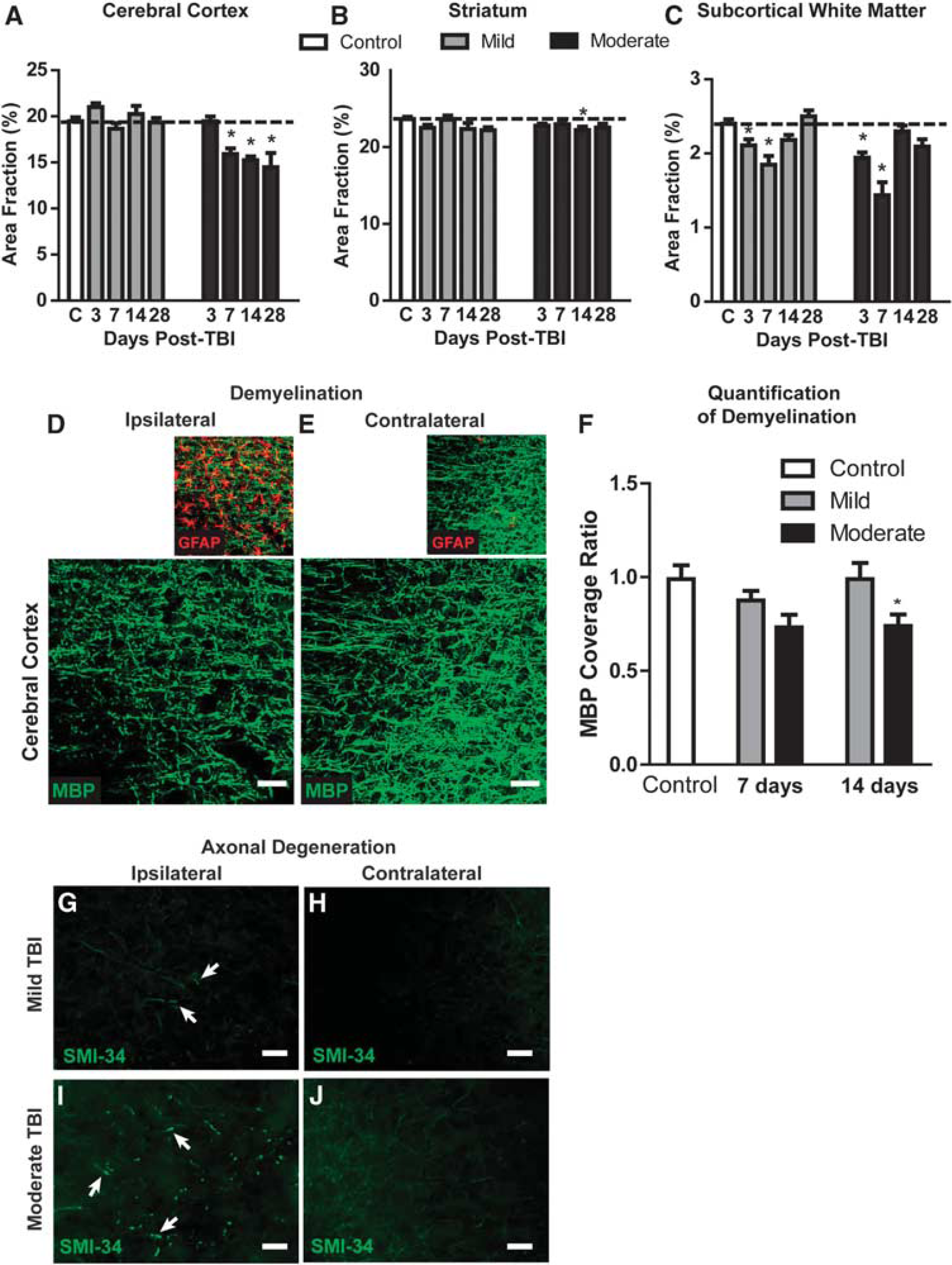
Axonal degeneration and demyelination after mild and moderate traumatic brain injury (TBI). (
Evolution of AQP4 Dysregulation After Mild and Moderate TBI
We next investigated how cortical and striatal AQP4 expression changes at 3, 7, 14, and 28 days after mild and moderate TBI (3–6 animals per group, four slices per animal, six image sets per slice). After immunofluorescence double labeling, GFAP and AQP4 expression were evaluated by image analysis. Representative images depicting cortical AQP4 expression under control conditions, and 7 days after mild and moderate TBI are shown in Figures 5A–5C while striatal AQP4 expression is shown in Figures 5G–5I. In prior studies, AQP4 expression was most commonly evaluated globally, either by Western blot14–21 or by grading of immunohistochemical staining after TBI.21–24 We first quantified changes in global AQP4 expression within the cortex and striatum after mild and moderate TBI. After both mild and moderate TBI, a slight increase in global AQP4 expression was apparent both in the cortex (Figure 5D) and in the striatum (Figure 5J) that appeared to peak 7 days post injury, then subside over the subsequent 3 weeks. These changes, however, did not reach statistical significance compared with control values.
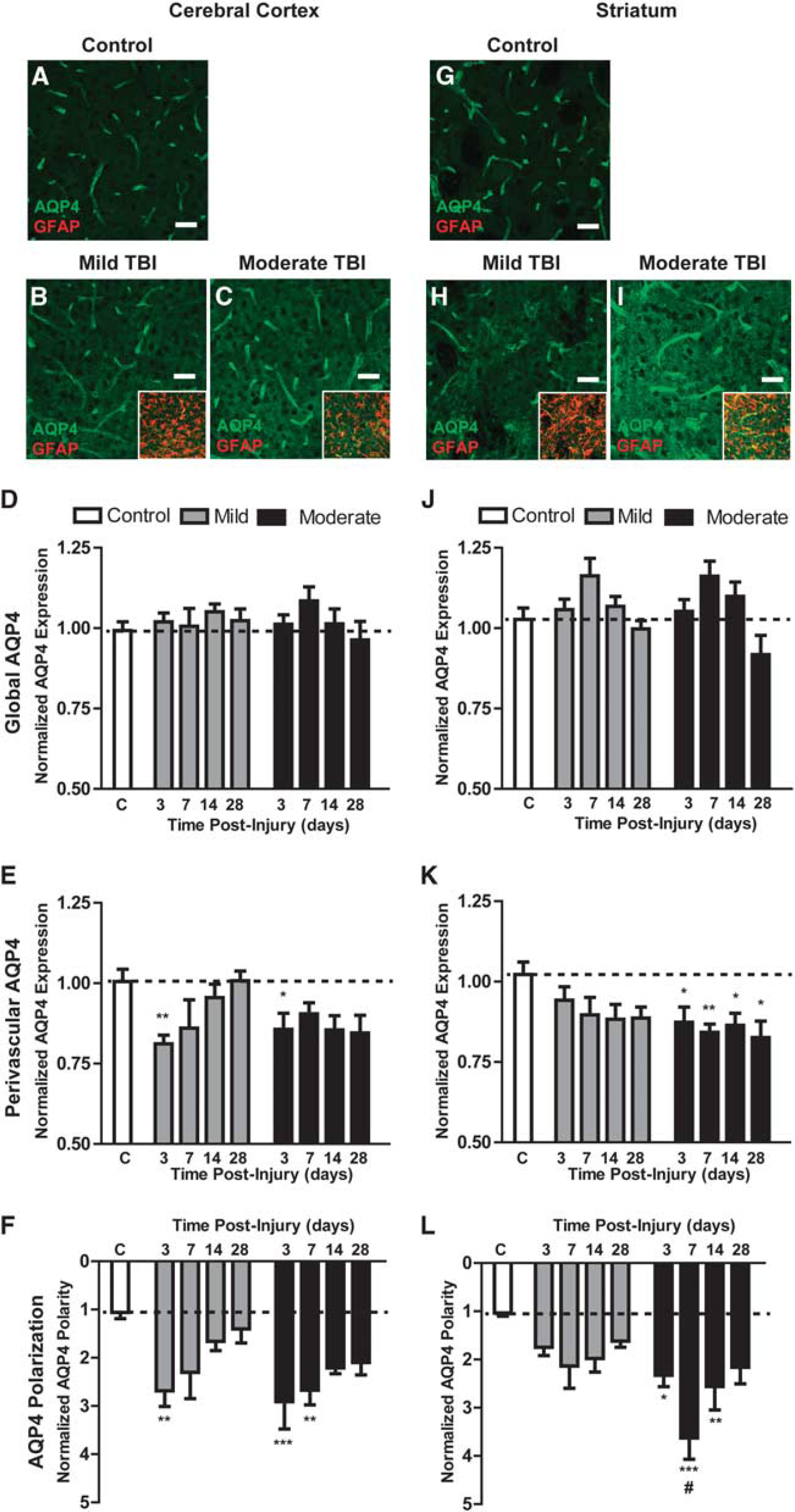
Evolution of post-traumatic AQP4 dysregulation. Astroglial AQP4 expression was evaluated by immunofluorescence in control cortex (
Because under control conditions AQP4 expression is restricted predominantly to perivascular astrocytic endfoot domains, a minority of prior studies have focused on changes in perivascular AQP4 expression after TBI.21–23,25 We next quantified posttraumatic changes in perivascular AQP4 expression in the cortex and striatum after mild and moderate TBI. In general, changes in perivascular AQP4 expression were more dramatic than those observed in global AQP4 expression. After mild TBI, cortical perivascular AQP4 expression was markedly reduced 3 days post injury (Figure 5E, ∗∗
Recently, Fukuda
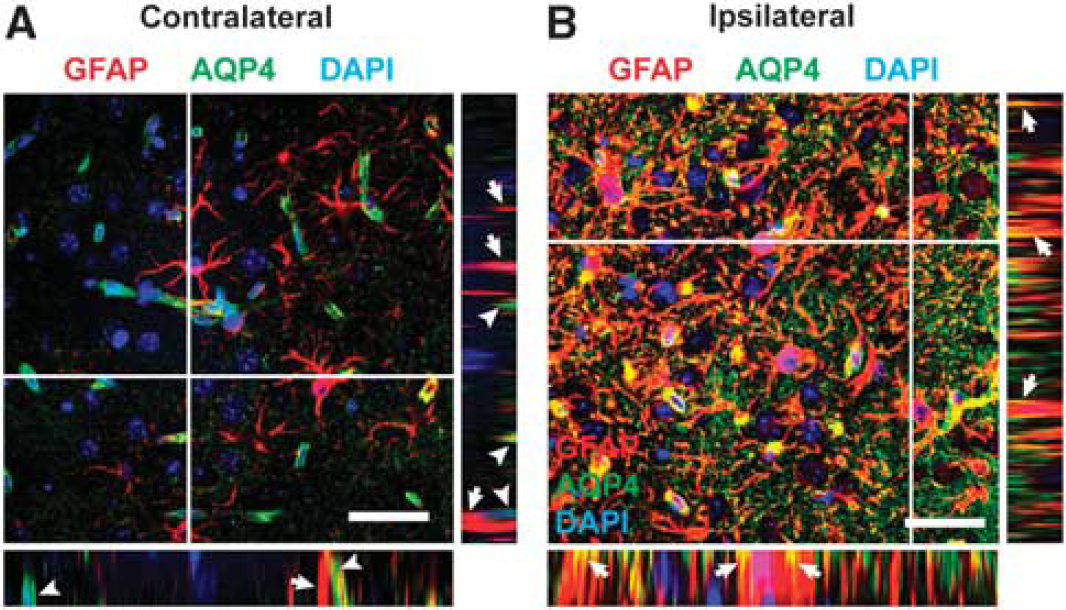
Post-traumatic redistribution of AQP4 localization. Localization of AQP4 was assessed by high-power confocal microscopy 14 days after moderate TBI. (
Based on these data, it is apparent that AQP4 expression and localization are undergoing complex changes within the cortex and striatum after TBI. A mild increase in global AQP4 expression coincided with a marked decrease in perivascular AQP4 expression. These two elements are apparently reflected in the striking shift in AQP4 polarization that we observed. To determine whether the changes in AQP4 expression vs. this shift in AQP4 localization were more robustly related to the onset of reactive astrogliosis, we performed a linear regression analysis comparing the extent of cortical and striatal GFAP expression, global and perivascular AQP4 expression, and AQP4 polarization at 3, 7, 14 and 28 days after mild and moderate TBI. The complete results of this analysis are presented in Supplementary Figure S2 and Supplementary Table S1. Significant differences between the regression lines were measured in the cortex 14 and 28 days post injury, and in the striatum at 3, 14, and 28 days post injury. At each of these time points, the slopes of the AQP4 polarization/ GFAP area fraction regression lines were significantly non-zero, with goodness of fit (
Moderate Hit & Run TBI resulted in frank disruption of cortical tissue local to the impact site that was evident 3 days after injury (Figure 3G), followed by the formation of a glial scar surrounding the lesion cavity (Figures 7C, 7E, and 7G). As the glial scar evolved and consolidated, marked changes in corresponding AQP4 expression were observed. Three days post injury, both GFAP and AQP4 labeling were globally increased, possibly reflecting increased non-specific antibody labeling in the disrupted tissue (Figures 7C and 7D). At 7 days post injury and continuing through 14 days post injury, a shift in AQP4 localization occurred that differed from that observed in the broader gliotic cortex or striatum. Proximate to the defined glial scar (Figures 7E and 7G), perivascular AQP4 immunoreactivity declined dramatically while AQP4 was massively upregulated in the consolidating glial scar (Figures 7F and 7H). This dramatic increase in AQP4 immunoreactivity drove the elevated overall AQP4 levels detected by image analysis (Figure 7B, ∗∗∗
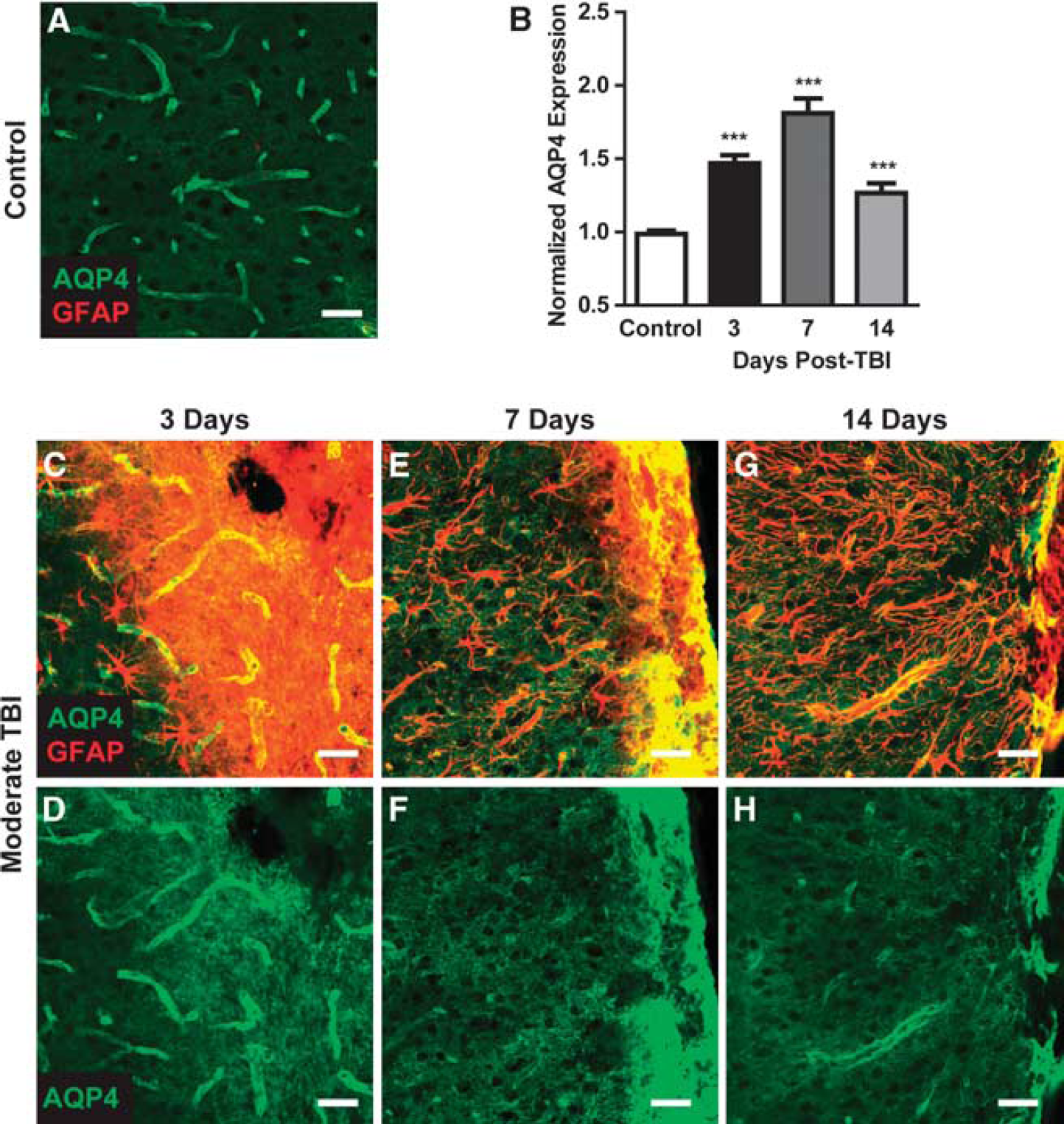
AQP4 expression in the developing glial scar. AQP4 expression was evaluated within control cortex (
DISCUSSION
We have developed and characterized a new TBI model, termed ‘Hit & Run’ TBI, in which (1) the mouse head and body are freely mobile during impact; (2) no pre-injury surgery is required; (3) the mice are anesthetized for only 2–3 minutes before impact; (4) the skull remains closed and skull fractures are avoided; and (5) the severity of injury can be modulated, including production of mild insult characterized by diffuse gliosis and white matter degeneration.
While numerous experimental TBI models have been developed in a variety of species including cats, dogs, and lower primates, rodent models have been widely adopted.1–3 One commonly used model is based on the gravitational forces of a free-falling weight to produce focal26,27 or diffuse28,29 brain injury through an impact delivered to the skull or the intact dura. The cortical contusion model30,31 utilizes a pneumatic piston to laterally deform the exposed dura. The fluid percussion injury model32,33 causes brain injury by rapidly injecting fluid volumes directly into the intact dural surface through a craniotomy.
The prolonged general anesthesia generally used in each of these preparations represents a key experimental shortcoming, as anesthetic agents can alter the pathophysiology of secondary injury.
34
In the Hit & Run model, the duration of anesthesia before impact is ~2.5 minutes, while animals are aroused from anesthesia between 1–3.5 minutes after impact. It is because the animals are awake and moving around their cage so rapidly after induction and impact that this model was termed ‘Hit & Run’ TBI. A second important element of this model is the fact that it is a closed-skull injury. A key pathologic feature of TBI and a chief driver of morbidity is cerebral edema and the consequent elevation in ICP that accompanies acute injury.
35
Cytotoxic cerebral edema results from the ischemic collapse of ionic gradients across the glial plasma membrane, facilitated by the activation of ion transporters such as NKCC1,
36
which in turn leads to BBB disruption, vasogenic edema formation, and intracranial hypertension. In cortical impact models in which the skull is opened to allow delivery of an impact directly to the cortical surface, ICP may be normalized as the cranial cavity is opened to the atmosphere. This likely alters the clinical course of secondary injury after traumatic insult. This is clearly illustrated in one study in rats evaluating the effect of craniotomy on cerebral edema and AQP4 expression after moderate TBI. In this study, enlarging the craniotomy beyond the size used for injury induction reduced both cerebral edema and AQP4 48 hours after injury.
15
This finding has important implications for the evaluation of changes in AQP4 expression after TBI and their role in post-traumatic edema formation. Without exception, all prior
In the present study, we use the Hit & Run TBI model to evaluate the relationship between cerebral edema, BBB disruption, changes in ICP, and AQP4 dysregulation after closed-skull traumatic injury for the first time. Our findings reveal a striking complexity in the evolution of AQP4 expression and localization from the acute (3 days) to the chronic phase (14–28 days) that may shed light on apparent contradictions within the existing literature. Prior studies, which have generally evaluated global AQP4 expression by western blot or gross scoring of immunohistochemical staining, have alternately reported that moderate TBI causes both reduced16,17,20,23 and elevated14,15,18,19,21–25,37 AQP4 expression. In the present study, we observed both elevations and reductions in AQP4 expression depending upon the tissue and the element of the tissue subjected to analysis. When perivascular AQP4 expression was evaluated within the cortex or the striatum (Figures 5E and 5K), a significant reduction in AQP4 expression was observed that persisted for more than 28 days after moderate TBI. In contrast, when global AQP4 expression was quantified, a slight increase was observed that peaked 7 days post injury (Figures 5D and 5J), then resolved by 28 days post injury. It is important to note that the regions subjected to this analysis included gliotic cortex and striatum, but excluded regions immediately adjacent to the lesion site and the defined glial scar. Within the glial scar itself, AQP4 immunoreactivity was intensely elevated. Thus, whether increased or decreased AQP4 expression is observed after TBI may depend largely upon whether global vs. perivascular AQP4 is being measured, and whether glial scar tissue is included in the analysis.
Prior studies focusing upon AQP4 after TBI have suggested that changes in AQP4 expression may drive edema formation and that reducing AQP4 expression may be a potentially useful therapeutic target.15,18,19,39 As mentioned above, these studies have been universally conducted in open-skull TBI models. In the present study, we report that ICP, BBB disruption, and cerebral edema peak 1–3 days post injury and largely normalize by 7 days post injury. In contrast, AQP4 dysregulation (including elevations in global AQP4 expression and AQP4 expression in the glial scar) increases up to 7 days post injury, only beginning to normalize in subsequent weeks. These findings suggest that AQP4 dysregulation may not drive cerebral edema formation within the acute phase. Rather, they are consistent with the notion that changes in AQP4 expression and localization may represent a compensatory mechanism to counteract cerebral edema and changes in ICP.
The present study is also the first to define changes in AQP4 expression and localization after mild TBI. Mild TBI is a common event, often resulting from sports injury, motor vehicle accident or falls, and blast exposure in the military. It is defined by the absence of structural brain injury by conventional neuroimaging, brief or absent loss of consciousness, transient changes in mental state or post-traumatic amnesia, and a Glasgow Coma Score ≥ 13. 40 While patients typically recover within 6 months of injury, more recent findings suggest that mild TBI can result in chronic diffuse axonal degeneration and neurodegenerative changes in the injured brain.41,42 In the Hit & Run model of mild TBI, we observed transient white matter atrophy and evidence of diffuse cortical axonal degeneration in the absence of cortical or striatal disruption or atrophy. Our behavioral analysis demonstrated that in contrast to moderate TBI animals, which exhibited both chronic cognitive (as measured by the novel object recognition and Barnes maze tests) and motor deficits (as measured by the rotarod test), animals undergoing mild TBI exhibited no detectable cognitive deficit but did exhibit a deficit in rotarod performance that was indistinguishable from that observed after moderate TBI. Mild injury resulted in cortical and striatal reactive astrogliosis (Figures 3D–3F) and microgliosis that peaked at 7 days post injury (Supplementary Figures S1A and S1B), then resolved between 14–28 days post injury. Persistent reactive astrogliosis was apparent in the ipsilateral hippocampus even 28 days post injury (Figure 3O). This is in contrast to moderate TBI, in which reactive gliosis in all regions persisted for at least 28 days post injury. In the acute phase (3–7 days), cortical AQP4 dysregulation after mild TBI was indistinguishable from that of moderate TBI, in terms of both reduced perivascular AQP4 expression (Figure 5E) and loss of AQP4 polarization (Figure 5F). In the chronic phase after mild TBI, cortical AQP4 expression and localization largely normalized by 28 days post injury. This is in contrast to APQ4 after moderate injury, which remained dysregulated at least 28 days post injury. Thus, in the first 7 days after injury, reactive astrogliosis and AQP4 dysregulation after mild and moderate TBI are largely indistinguishable. However, within the chronic phase, reactive astrogliosis and AQP4 dysregulation after mild and moderate TBI follow dramatically different courses.
Under physiologic conditions, AQP4 expression in the brain is localized to perivascular astrocytic endfeet and the glial limitans. The physiologic function of this highly polarized pattern of expression remains unclear, but may include the regulation of extracellular volume and K+ in response to neuronal activity43,44 and the maintenance of paravascular bulk flow through the brain parenchyma. 45 In either case, the polarization of AQP4 expression is likely a key determinant of proper AQP4 function, a notion supported by findings in mice harboring the targeted disruption of the gene encoding α-syntrophin. This scaffolding protein is necessary for AQP4 anchoring at the perivascular endfoot membrane and in this knockout mouse, although overall AQP4 expression is not altered, AQP4 localization is shifted away from the endfoot domains,44,46 resulting in impaired AQP4-dependent extracellular K+ regulation 44 and reduced formation of cerebral edema after cerebral ischemia. 46
The highly specific localization of AQP4 suggests that under pathologicconditions, changes in the relative polarization of this protein to the perivascular astrocytic endfeet may be more consequential than changes in its overall expression. Prior studies have used western blot of brain homogenate14–21 and gross scoring of immunohistochemical AQP4 labeling21–24 to measure changes in global or perivascular AQP4 expression after moderate TBI, while none have quantified changes in AQP4 polarization after TBI. Yet when we evaluated the relationship between AQP4 and reactive astrogliosis after mild and moderate TBI (Supplementary Figure S2 and Supplementary Table S1), we found that alterations in AQP4 polarization, particularly in the chronic phase, 14 and 28 days post TBI, were significantly related to the extent of reactive astrogliosis. Here, it is important to note that loss of AQP4 polarization to the perivascular endfeet was observed after both mild and moderate TBI and that mild TBI did not result in any apparent BBB disruption of cerebral edema. This suggests that loss of perivascular AQP4 polarization may be a general feature of reactive astrocytes, a notion supported by our report of similar chronic loss of AQP4 polarization in reactive astrocytes after diffuse microinfarction. 47 Whether such changes in AQP4 polarization represent adaptive response to brain injury or whether these changes drive secondary injury progression remain unclear. Inhibition of AQP4-dependent water flux into perivascular astrocytes may limit cytotoxic edema formation, 9 yet may similarly inhibit the resolution of vasogenic cerebral edema.13,48 Our recent report that AQP4-dependent paravascular bulk flow supports the clearance of soluble amyloid β from the brain 45 raises the additional possibility that chronic loss of perivascular AQP4 polarization may retard interstitial waste removal and thus may have unappreciated chronic consequences. Within this framework, targeting reactive gliosis in the chronic phase may provide a therapeutic avenue to normalize AQP4 expression and improve clearance of interstitial wastes after moderate or severe TBI. The present model of TBI, by avoiding head fixation, prolonged anesthesia, and craniotomy represents a useful tool in evaluating the development of chronic TBI-associated outcomes, including post-traumatic epilepsy and cognitive impairment, and may provide a straightforward approach to evaluating the role of single and repeated mild TBI in the development of chronictraumatic encephalopathy.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.