
Abstract
The organic cation transporters OCT1, 2, and 3 (SLC22A1-3) have been implicated in the elimination of biogenic amines such as histamine. Among them, OCT3 was identified as an uptake-2 transporter, responsible for clearance of histamine. Because increasing evidence suggests the involvement of histamine in cerebral ischemia, we investigated the effects of targeted disruption of organic cation transporter-3 (Oct3) on the severity of ischemic brain damage. Transient focal ischemia for 1 hour was induced by occlusion of the middle cerebral artery (MCA) of homozygous Oct3-deficient mice and their wild-type (Wt) littermates. Although targeted disruption of Oct3 did not affect physiological parameters after MCA occlusion, this disruption significantly increased histamine content in the ischemic cortex and significantly reduced the infarct volume after cerebral ischemia. Furthermore, targeted disruption of Oct3 prevented the reduction of regulatory T-cell proportion after cerebral ischemia while this disruption did not affect Th1 and Th2 cells proportions after ischemia. Since repeated administration of
Introduction
Cerebral ischemia results from a decrease in regional cerebral blood flow (CBF) sufficient to trigger a complex cascade of cellular events such as excitotoxicity, disrupted calcium homeostasis, peri-infarct depolarization, oxidative and nitrative stress, mitochondrial dysfunction, apoptosis, and inflammation (Doyle et al, 2008; Durukan and Tatlisumak, 2007; Hossmann, 2006). Among these events, there have been major advances in the study of the role of inflammation after cerebral ischemia. There is increasing evidence that inflammation and immune response have an important role in the progression of ischemic brain injury (Brea et al, 2009; Denes et al, 2010).
Histamine is a biogenic amine with multiple physiological activities. Histamine is known to participate in allergic and inflammatory reactions, immunomodulation, gastric acid secretion, and neurotransmission, which are mediated through four different G protein-coupled receptor subtypes (H1 to H4; Huang and Thurmond, 2008). In the immune system, histamine is not only an important proinflammatory mediator of immediate type allergic responses, but it can also modulate inflammatory responses and affect chronic inflammation (Jutel et al, 2009). The histamine H1 receptor (H1R) is thought to be responsible for acute inflammatory responses. It stimulates immune system cells by potentiating their proinflammatory activity to increase migration to the area of inflammation as well as increase effector functions. In contrast, the histamine H2 receptor (H2R) seems to be a potent suppressor of inflammatory and effector functions. Histamine H4 receptor (H4R) activation promotes the accumulation of inflammatory cells at sites of allergic inflammation (Huang and Thurmond, 2008). It is, therefore, generally accepted that histamine has a pivotal role in modulating the progression of ischemic brain injury. In fact, it has been reported that repeated administration of
The organic cation transporters 1, 2, and 3 (OCT1-3, namely SLC22A1-3) mediate the facilitated transport of diverse organic cations, including many drugs, toxins, and endogenous compounds. Besides a possible role of the OCT transporters in the clearance of xenobiotics, they have also been implicated in the elimination of biogenic amine neurotransmitters such as tyrosine-derived catecholamines (dopamine, epinephrine, and norepinephrine), serotonin (5-hydroxytryptamine), and histamine (Jonker and Schinkel, 2004). Among them, OCT3 was identified as the uptake-2 transporter in diverse tissues such as the kidney, heart, vascular system, and central nervous system (Zwart et al, 2001). In addition, it was reported that OCT3 is the molecule responsible for uptake and clearance of histamine in tissues (Ogasawara et al, 2006; Schneider et al, 2005). For these reasons, we speculated that OCT3 could be involved in the mechanism of cerebral ischemia via the control of histamine level. To confirm our assumption, we examined the influence of targeted disruption of organic cation transporter-3 (Oct3) on ischemic brain damage using a transient middle cerebral artery occlusion (MCAO) model in mice. Here, we show that targeted disruption of Oct3 ameliorated ischemic brain damage by modulating the histamine content and the Treg population in mice.
Materials and methods
All experiments were approved by the Ethics Committee of Ehime University Graduate School of Medicine and were conducted according to the Guidelines for Animal Experimentation at Ehime University Graduate School of Medicine.
Animals
Adult male homozygous Oct3 knockout (Oct3 KO) mice (Zwart et al, 2001) and their wild-type (Wt) littermates, 10 to 12 weeks old, were used in this study. All mice were of FVB genetic background.
Transient Middle Cerebral Artery Occlusion
Both Oct3 KO mice and Wt littermates were anesthetized with 1.5% halothane in a 4:3 mixture of nitrous oxide and oxygen. An 8-0 nylon monofilament (Ethilon; Ethicon, Norderstedt, Germany) coated with silicon resin (XantoprenM; Bayer Dental, Osaka, Japan) was introduced through a small incision into the common carotid artery and advanced 9 mm distal to the carotid bifurcation for occlusion of the middle cerebral artery (MCA) (Hata et al, 1998). Sham operation was performed by insertion of a thread into the common carotid artery, without advancing it to occlude the MCA. At 1 hour after occlusion, the thread was removed to allow reperfusion of the MCA territory. Rectal temperature was maintained at 37.0±0.5°C during occlusion and until 1 hour after reperfusion. After confirming recovery from anesthesia, animals were maintained in an air-conditioned room at ∼22°C.
2,3,5-Triphenyltetrazolium Chloride Staining
At 24 hours after reperfusion, both Oct3 KO mice and Wt littermates were killed under deep anesthesia with a lethal dose of sodium pentobarbital (0.1 g/kg). Their brains were removed and sectioned coronally into 1 mm slices using a mouse brain matrix (BRM-2000C; Activational System Inc., Warren, MI, USA), and immediately stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma, St. Louis, MO, USA) and incubated at 37°C for 30 minutes. The border between infarcted and noninfarcted tissue was outlined using NIH image software (National Institutes of Health, Bethesda, MD, USA), and the area of infarction was measured by subtracting the area of the lesioned hemisphere from that of the nonlesioned contralateral hemisphere (Swanson et al, 1990). The volume of infarction was calculated by integration of the lesion areas at seven equidistant levels of the forebrain.
Cerebral Angioarchitecture
Both Oct3 KO mice and Wt littermates were deeply anesthetized with a lethal dose of sodium pentobarbital (0.1 g/kg). Warm (38°C) undiluted latex (Vultex; Chicago Latex, Chicago, IL, USA) mixed with carbon black (10 μL/mL, Bokusai; Fueki, Tokyo, Japan) was injected transcardially. The injected volume of latex was 0.4 mL and injection pressure was ∼150 mm Hg. Thereafter, the whole head was fixed in 10% formalin for 4 weeks before brain removal. Anastomoses on the dorsal surface of the hemispheres were localized by tracing the peripheral branches of the anterior cerebral artery and the MCA to the anastomosis points. Adjacent anastomosis points were connected by the line of anastomoses (Maeda et al, 1998). The distance from the midline to the line of anastomoses was measured in coronal planes 2, 4, and 6 mm from the frontal pole in photographs taken from the dorsal brain surface, using NIH Image software (National Institutes of Health).
Cerebral Blood Flow
Cerebral blood flow was determined in the territory of MCA by laser-Doppler flowmetry (LDF) using a flexible 0.5-mm fiberoptic extension to the masterprobe (FLO-N1; Neuroscience Inc., Tokyo, Japan). In both Oct3 KO mice and Wt littermates, the tip of the probe was fixed to the intact skull over the territory supplied by the proximal part of the MCA (2 mm posterior and 6 mm lateral to the bregma) using a tissue adhesive (Aron Alpha, Toa, Tokyo, Japan). Changes in cortical perfusion after MCAO were expressed as a percentage of the baseline value of LDF.
Histamine Measurement
Tissue histamine level was measured by high-performance liquid chromatography-fluorometry as described by Yamatodani et al (1985). The high-performance liquid chromatography system included a cation exchange column (TSK-gel SP-2SW; 6 mm intradermally × 150 mm, Tosoh, Tokyo, Japan) to separate histamine. Elutants were then mixed with 0.1% o-phthaldialdehyde and 2 mol/L NaOH in a reaction coil made of polytetrafluoroethylene tubing (0.5 mm intradermally × 5 m) at 45°C and pH 12.0, and finally, 10% of sulfuric acid was added to reduce the pH to 3.0. o-Phthaldialdehyde, NaOH, and sulfuric acid solutions were pumped out at 0.2 to 0.25 mL/min. Fluorescence was monitored using a fluorescent detector (L-7480; Hitachi, Tokyo, Japan) using excitation and emission wavelengths of 360 and 450 nm, respectively.
Cytokine Measurement
Tissue MCP-1, IL-6, TNF-α, INF-γ, IL-10, and IL-12 concentrations were measured with a CBA Mouse Inflammation Kit according to manufacturer's instructions (BD Biosciences, San Jose, CA, USA). A mixture of six capture bead populations, each with distinct fluorescence intensities and coated with antibodies specific for IL-6, IL-10, MCP-1, IFN-γ, TNF-α, and IL-12p70, were prepared. The CBA capture beads were incubated together with phycoerythrin (PE)-conjugated detection antibodies, test samples or standards, to form sandwich complexes. After acquisition of sample data using FACScan flow cytometer (FACS Calibur; BD Biosciences), the sample results were generated in graphical and tabular format using the CBA Analysis Software (BD Biosciences).
Flow Cytometric Analysis
The mouse was killed under deep anesthesia with a lethal dose of sodium pentobarbital (0.1 g/kg). Blood samples (1.0 to 1.5 mL from each mouse) were collected in heparin-rinsed syringes for the isolation of peripheral blood mononuclear cells. After determining the viability of cells by trypan blue exclusion method, the cells were resuspended in a cell staining buffer. The freshly prepared cells were analyzed using direct immunofluorescent staining. Approximately 106 cells of each sample were incubated for 30 minutes on ice in cell staining buffer (Biolegend, San Diego, CA, USA) containing monoclonal antibodies. For analysis of Th1 and Th2 cells, the cells were labeled in cell staining buffer containing PE/Cy5-conjugated antimouse CD4 (Biolegend). Then, cells were incubated with FITC-conjugated antimouse IFN-γ or PE-conjugated antimouse IL-4. For analysis of Treg, cells were labeled in cell staining buffer containing APC-conjugated antimouse CD4 and PE-conjugated antimouse CD25 (Biolegend). For intracellular labeling, cells were fixed and washed with FOXP3 fix.perm.buffer, perm.washing.buffer and FOXP3 perm.buffer (Biolegend). Cells were then incubated with Alexa fluor-488-conjugated antimouse FOXP3 antibodies (Biolegend). After the final wash, the cells were resuspended in cell staining buffer and the cells were analyzed by cytometry using a FACScan flow cytometer (FACSCalibur; BD Biosciences). The importance and accuracy of this method for measurements of Tregs were described elsewhere (Sakaguchi, 2005).
Repeated Administration of L -Histidine
Both Oct3 KO mice and their Wt littermates were divided into two groups, control and treated group. A total of 400 μL
Statistical Analysis
All values are presented as mean±s.d. Differences in infarct volume and CBF reduction rate between Oct3 KO mice and Wt littermates were analyzed statistically by unpaired t test. All other statistical significance was tested by one-way ANOVA followed by Bonferroni's multiple comparison test. A P value <0.05 was considered statistically significant.
Results
Targeted Disruption of Oct3 Gene in Mice Reduced Infarct Volume after Transient Focal Ischemia
First, we investigated the effect of targeted disruption of Oct3 on the severity of ischemic brain damage. Transient focal ischemia was induced for 1 hour by occlusion of the right MCA in Oct3 KO mice as well as in their Wt littermates (n=6 in each group). Twenty-four hours after reperfusion, TTC staining showed a significant reduction of infarct area at 3 and 4 mm from the frontal pole of the brain in KO mice compared with that in Wt littermates (Figure 1B). In KO mice, the total infarct volume was 39.4±14.3 (mean±s.d.) mm3, while it was significantly higher at 77.0±20.8 mm3 in Wt littermates (Figure 1C).
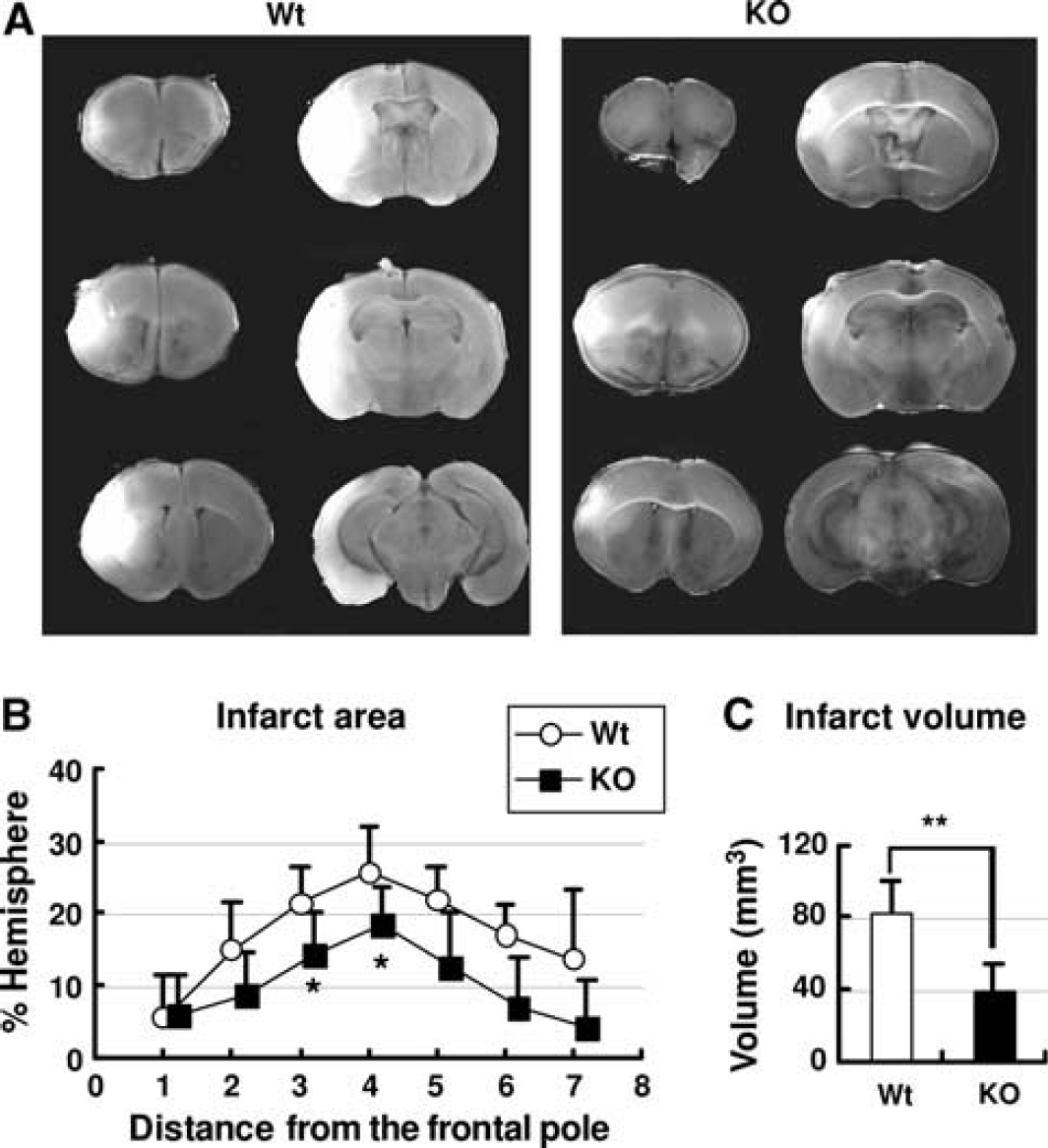
Targeted disruption of organic cation transporter-3 (Oct3) reduced infarct volume. Transient focal ischemia for 1 hour was induced by occlusion of the right middle cerebral artery in Oct3 knockout mice (KO; n=6) as well as in their wild-type littermates (Wt; n=6). Brain infarcts were visualized by 2,3,5-triphenyltetrazolium chloride (TTC) staining 1 day after reperfusion in six coronal brain slices prepared at 1 mm intervals from the frontal pole (
Measurement of Physiological Parameters and Semiquantitative Evaluation of Cerebral Angioarchitecture and Cerebral Blood Flow Showed No Difference between Oct3-Deficient Mice and Their Wild-Type Littermates
We next evaluated the physiological parameters (mean arterial blood pressure, heart rate, PaCO2 etc.) of KO mice and Wt littermates just before MCAO and one day after MCAO, showing that there was no significant difference in each parameter between the two groups (n=4 in each group, see Supplementary Tables 1 and 2).
To exclude the possibility that the reduced infarct volume in KO mice was caused by different angioarchitecture from that in Wt littermates, we used latex mixed with carbon black to evaluate the brain angioarchitecture. The anastomosis points of the anterior cerebral artery and MCA were marked to show the MCA-supplied area (Figure 2A). As shown in Figure 2B, the distance of the line of anastomoses from the midline showed no significant difference between KO mice and Wt littermates (n=6 in each group).
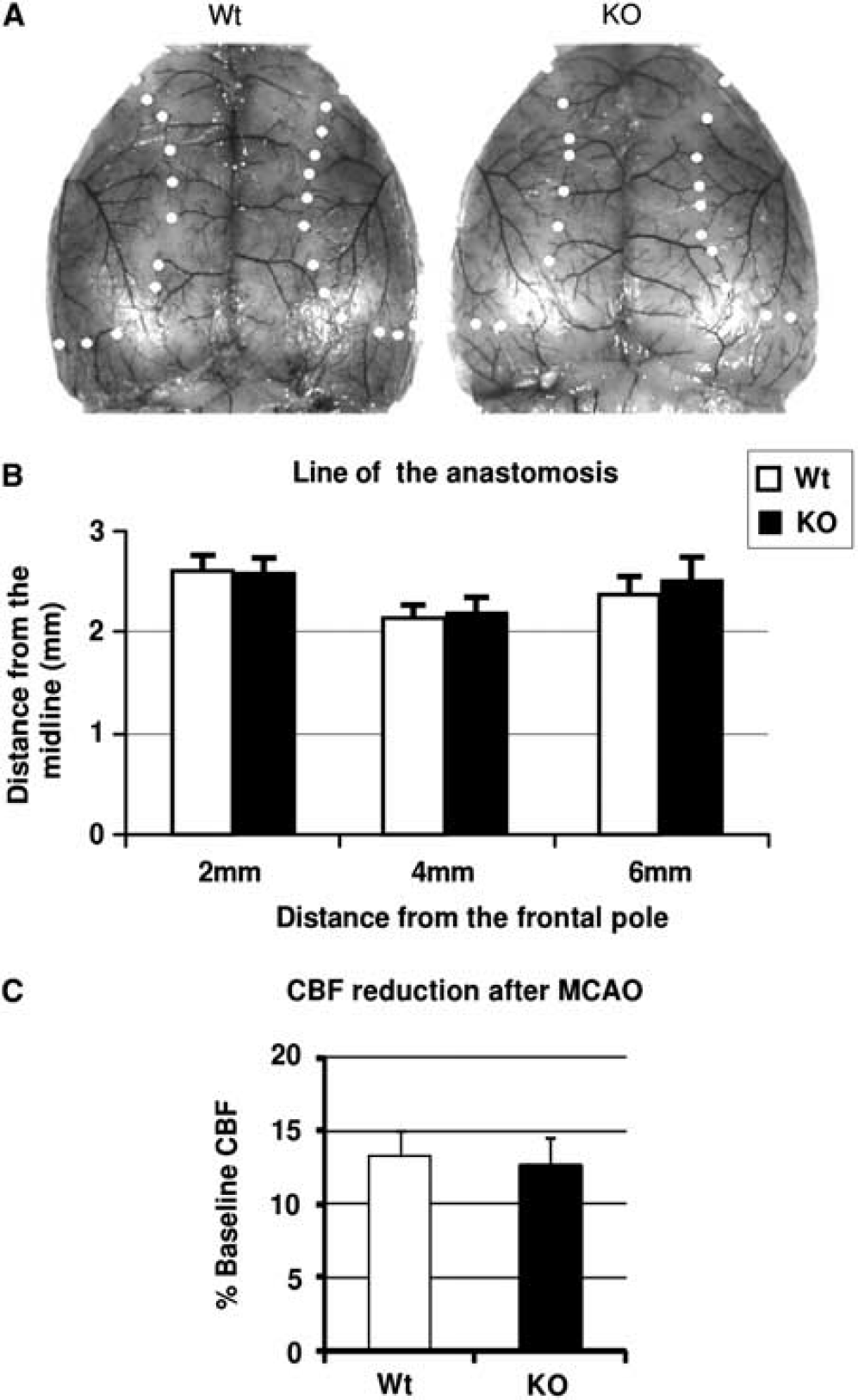
Brain vasculature and cerebral blood flow (CBF) reduction after middle cerebral artery occlusion (MCAO). Both organic cation transporter-3 (Oct3) knockout (KO) mice and wild-type (Wt) littermates were deeply anesthetized with a lethal dose of sodium pentobarbital (0.1 g/kg). Warm (38°C) undiluted latex (0.4 mL Vultex; Chicago Latex) mixed with carbon black (10 μL/mL, Bokusai; Fueki) was injected transcardially. Thereafter, the whole head was fixed in 10% formalin for 4 weeks before brain removal. The points of anastomoses between the middle and the anterior cerebral arteries are circled and connected by the line of anastomoses to demarcate the respective vascular territories in the dorsal brain surface of an Oct3 knockout (KO) mouse and a wild-type (Wt) littermate (
Moreover, to exclude the possibility that the difference in infarct volume was caused by different residual CBF during and after ischemia, we measured CBF using laser-Doppler flow (LDF). Immediately after MCAO, CBF decreased in the parietal cortex to ∼12% of baseline (Wt: 13.3±1.78% (mean±s.d.); KO: 12.4±2.13%, n=5 in each group) and remained at this level throughout the 1 hour of vascular occlusion. During ischemia, no significant difference in residual CBF was found between KO mice and Wt littermates (Figure 2C). When the intraluminal thread was removed after 1 hour ischemia, CBF instantaneously rose and returned to the baseline within 10 minutes after reperfusion in all animals.
Taken together, these results suggest that the significant reduction of ischemic damage in Oct3-deficient mice was not related to either morphological or functional differences in the brain angioarchitecture between KO mice and Wt littermates.
Histamine Level after Transient Middle Cerebral Artery Occlusion
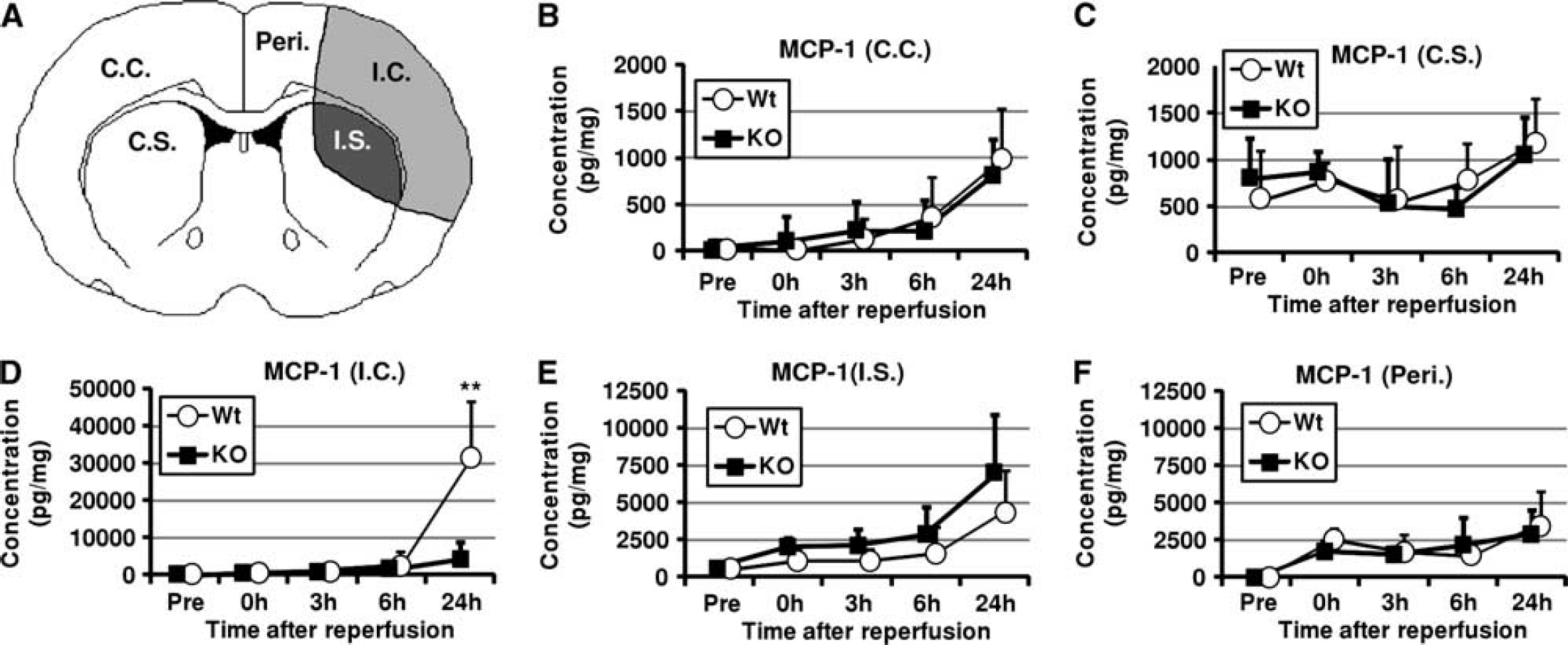
To investigate the influence of targeted disruption of Oct3 on histamine level after cerebral ischemia, we next measured the histamine level in the brain and serum before and after transient MCAO. As shown in Figure 3A, at 0, 3, 6, and 24 hours after reperfusion, tissue samples were taken from the control cortex (C.C.), control striatum (C.S.), ischemic cortex (I.C.), ischemic striatum (I.S.), peri-ischemic cortex (Peri.), and serum (SE.) in both Oct3 KO mice (n=5 at each time point) and Wt littermates (n=5 at each time point). Tissue samples taken from sham-operated animals (n=5 in each group) were used as control (Pre).
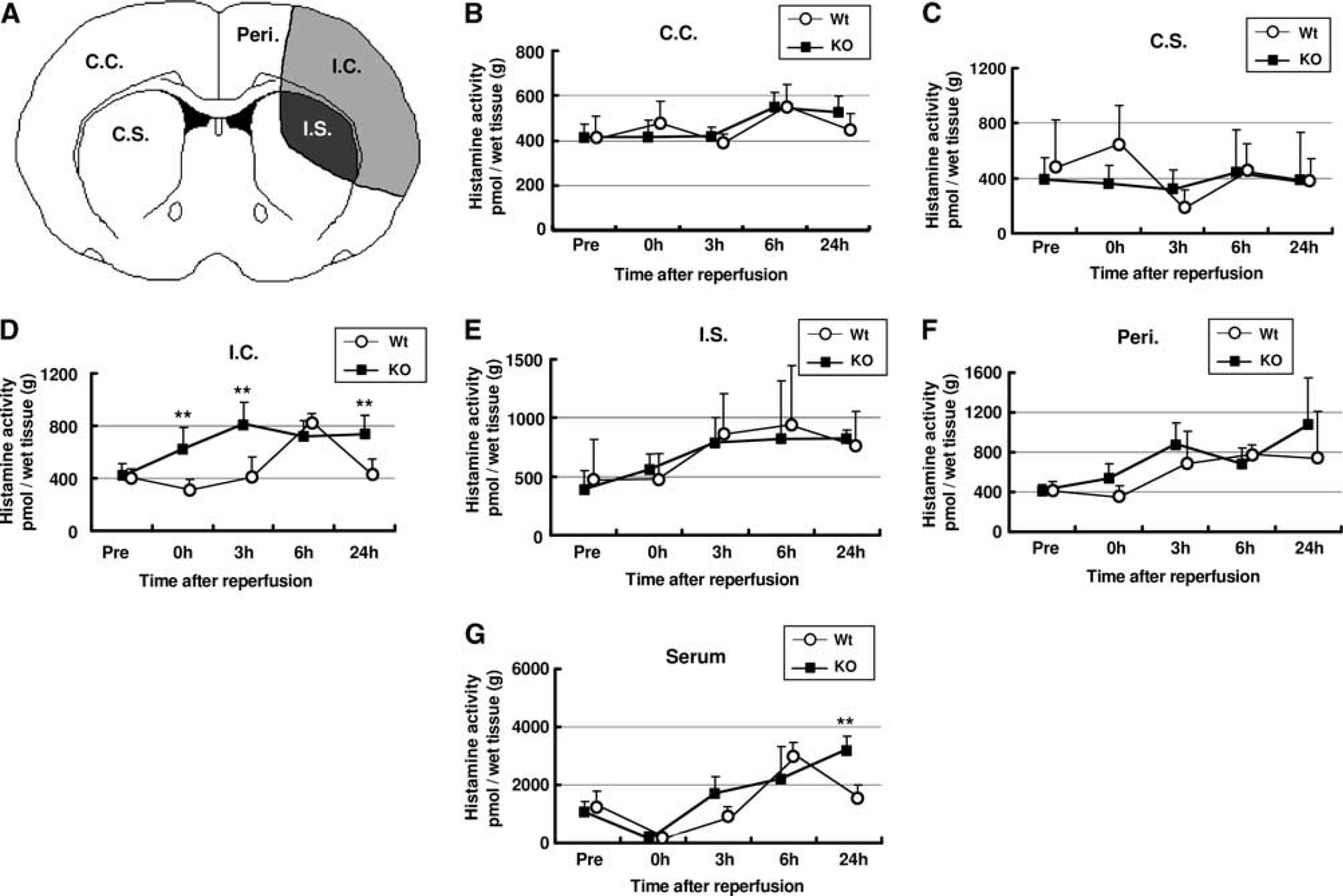
Effects of targeted disruption of organic cation transporter-3 (Oct3) on histamine level after cerebral ischemia. (
Before ischemia there was no significant difference in tissue histamine levels between KO mice and Wt littermates (Figure 3).
Histamine level in brain
Histamine level in the C.C. and C.S. showed no significant difference between KO mice and Wt littermates during ischemia and up to 24 hours after reperfusion (Figures 3B and 3C). In the I.C., histamine level increased during ischemia and after reperfusion in KO mice. It reached a peak at 3 hours after reperfusion (813±148 pmol/g), and was maintained at this level up to 24 hours after reperfusion, while in Wt littermates, during ischemia and until 3 hours after reperfusion, there was no significant difference in histamine level compared with the preischemia level (408±102 pmol/g). Histamine level started to increase 3 hours after reperfusion, peaked at 6 hours after reperfusion (831±43.4 pmol/g), and decreased to the preischemia level at 24 hours after reperfusion (415±123 pmol/g). Our results showed that in I.C., histamine level in KO mice was significantly higher than that in Wt littermates during the ischemic period, and 3 and 24 hours after reperfusion (Figure 3D). Histamine levels in I.S. and Peri. started to increase after reperfusion and remained increased up to 24 hours after reperfusion. There was no significant difference between these two animal groups (Figures 3E and 3F).
Histamine level in serum
In both KO mice and Wt littermates, serum histamine level decreased during ischemia. After the thread was removed, histamine level increased in both groups. In Wt littermates, serum histamine level peaked at 6 hours after reperfusion (2,990±460 pmol/mL) and decreased to the preischemia level at 24 hours after reperfusion (1,500±500 pmol/mL), while in KO mice, histamine level in serum increased until 24 hours after reperfusion (3,220±467 pmol/mL). Serum histamine level in KO mice was significantly higher than that in Wt littermates at 24 hours after reperfusion (Figure 3G).
Cytokine Level after Transient Middle Cerebral Artery Occlusion
Since tissue histamine level in KO mice was significantly higher than that in Wt littermates 24 hours after reperfusion, and histamine modulates cytokine and chemokine expression, we next evaluated the levels of IL-6, MCP-1, TNF-α, INF-γ, IL-10, and IL-12 before and after ischemia. At 0, 3, 6, and 24 hours after reperfusion, tissue samples were taken from the C.C. (see Figures 4A and 5A), C.S., I.C., I.S., and Peri. in both Oct3 KO mice (n=5 at each time point) and Wt littermates (n=5 at each time point). Tissue samples taken from sham-operated animals (n=5 in each group) were used as control (Pre).
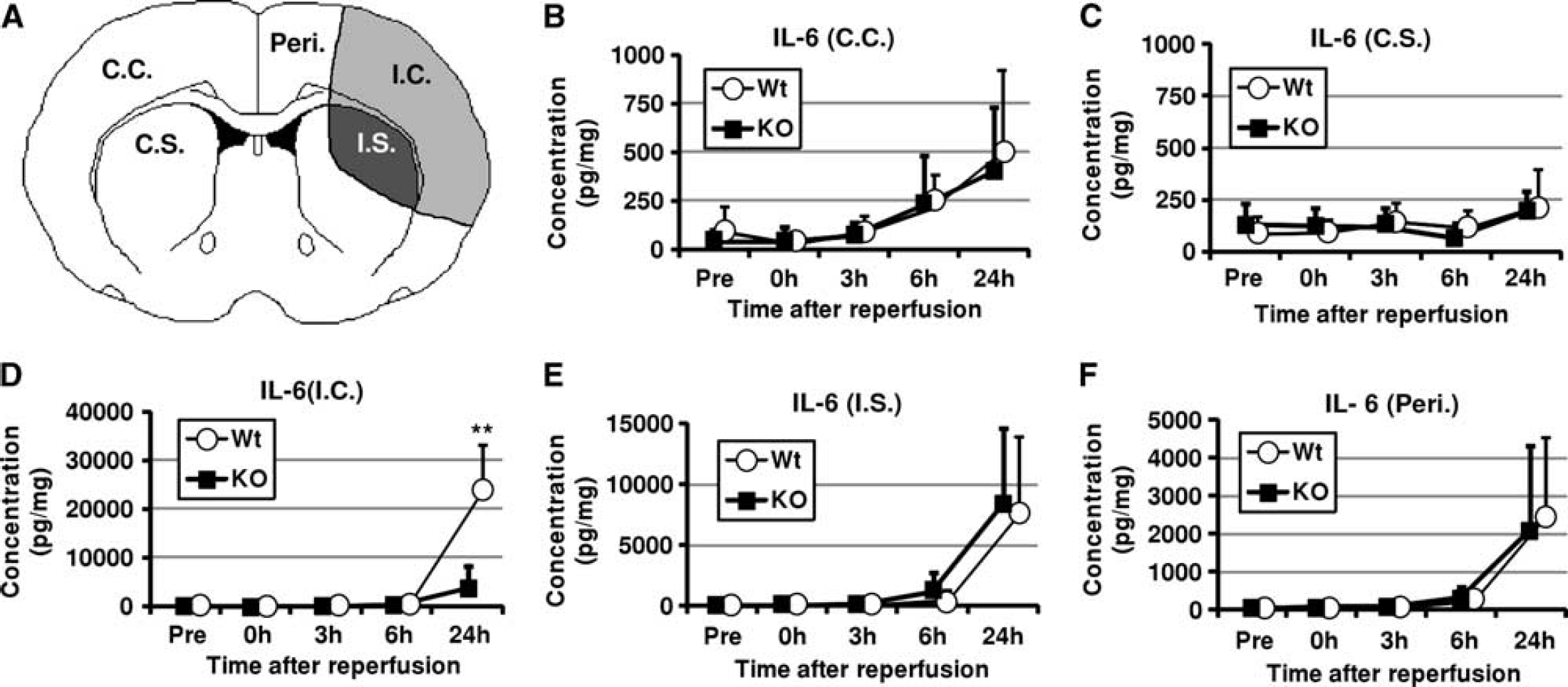
Effects of targeted disruption of organic cation transporter-3 (Oct3) on IL-6 level after cerebral ischemia. (
Effects of targeted disruption of organic cation transporter-3 (Oct3) on MCP-1 level after cerebral ischemia. (
As shown in Figures 4 and 5, the levels of IL-6 and MCP-1 in all areas of the brain were significantly increased at 24 hours after reperfusion. Especially, in the I.C., I.S., and Peri., the levels of IL-6 and MCP-1 were markedly increased in both groups. Among them, the levels of IL-6 and MCP-1 in I.C. of Wt littermates were significantly higher than those in KO mice at 24 hours after reperfusion ((IL-6) Wt: 23,500±9,560 pmol/mg (mean±s.d.); KO: 5,460±3,340 pmol/mg), ((MCP-1) Wt: 31,300±15,300 pmol/mg; KO 4,280±4,600 pmol/mg). As shown in Supplementary Figures 1 and 2, the level of TNF-α in I.C. gradually increased up to 24 hours after reperfusion (Supplementary Figure 1D), while the level of INF-γ in I.C. increased after reperfusion, peaked at 3 hours after reperfusion, and decreased but was maintained at higher levels than the preischemia level up to 24 hours after reperfusion (Supplementary Figure 2D). There were no significant differences in the levels of TNF-α and INF-γ between KO mice and Wt littermates. As shown in Supplementary Figures 3 and 4, there were no significant differences in the levels of IL-10 and IL-12p70 between KO mice and Wt littermates.
Cytokine and Nitric Oxide Secretion from Astrocytes, Microglia, and Macrophages after Activation by Lipid A
We next investigated the effects of targeted disruption of Oct3 on the secretion of IL-6, MCP-1, TNF-α, and nitric oxide (NO) in astrocytes, microglia, and bone marrow-derived macrophages. The levels of IL-6, MCP-1, TNF-α, and NO in the culture media of these three types of primary cultured cells were measured at 3, 6, 12, 24, and 72 hours after activation by Lipid A in the presence or absence of IFN-γ. Data were obtained from five independent measurements.
Without activation by Lipid A, IL-6, MCP-1, TNF-α, and NO could not be detected in the culture media up to 72 hours after incubation (data not shown).
At 3 or 6 hours after activation by Lipid A, IL-6, MCP-1, and TNF-α could be detected in the media of these three types of primary cultured cells. The levels of IL-6, MCP-1, and TNF-α significantly increased in a time-dependent manner. However, there were no significant differences in the levels of these cytokines between KO mice and their Wt littermates (Supplementary Figures 5 to 7). In astrocytes and microglia, NO could be detected at 24 hours (astrocytes) and 72 hours (astrocytes and microglia) after activation by Lipid A+INF-γ (Supplementary Figures 5D and 6D). In macrophages, NO could be detected at 24 and 72 hours after activation by Lipid A in the presence and absence of INF-γ (Supplementary Figure 7D). In all cases, there were no significant differences in the level of NO between KO mice and their Wt littermates (Supplementary Figures 5D to 7D).
Populations of Th1, Th2, and Treg Cells after Cerebral Ischemia
Our previous data showed that targeted disruption of Oct3 did not directly affect the secretion of IL-6, MCP-1, TNF-α, and NO in astrocytes, microglia, and macrophages. Since histamine can reciprocally regulate T-cell activity by H1 and H2 receptor activation, we next investigated the effects of targeted disruption of Oct3 on the proportions of T-cell subtypes after cerebral ischemia. At 3 and 24 hours after reperfusion, the proportions of Th1, Th2, and Treg cells in KO mice (n=5 at each time point) and their Wt littermates (n=5 at each time point) were measured by flow cytometric analysis. Sham-operated animals of both strains (n=5 in each group) were used as control (before ischemia, Pre).
Before ischemia (Pre), there were no differences in the proportions of Th1, Th2, and Treg cells between KO mice and their Wt littermates (Figure 6). The proportions of these three types of T cells were significantly decreased at 3 hours after reperfusion and recovered at 24 hours after reperfusion. There were no significant differences in proportions of Th1 and Th2 cells between KO mice and their Wt littermates (Figures 6A and 6B). However, the proportion of Treg cells in KO mice was significantly higher than that in their Wt littermates after reperfusion (Figure 6C).
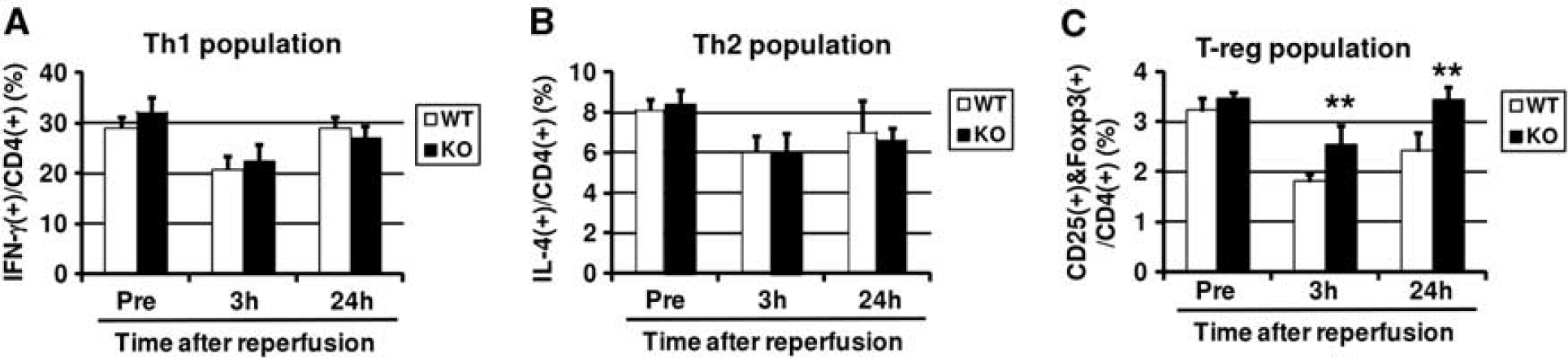
Effects of targeted disruption of organic cation transporter-3 (Oct3) on Th1, Th2, and Treg populations after cerebral ischemia. At 3 and 24 hours after reperfusion, blood samples were taken from both Oct3 knockout mice (KO; n=5 at each time point) and wild-type littermates (Wt; n=5 at each time point). Blood samples taken from sham-operated animals (n=5) were used as control (Pre). For analysis of Th1 cells and Th2 cells, the cells were labeled with phycoerythrin (PE)/Cy5-conjugated antimouse CD4, FITC-conjugated antimouse IFN-γ, and PE-conjugated antimouse IL-4 (Biolegend). For analysis of regulatory T cells, cells were labeled with APC-conjugated antimouse CD4, PE-conjugated antimouse CD25 and Alexa fluor-488-conjugated antimouse FOXP3 antibodies (Biolegend). The cells were analyzed by cytometry using a FACScan flow cytometer (FACSCalibur; BD Biosciences, Pasadena, CA, USA). Data were obtained from five independent measurements. Values represent mean±s.d. Note that the proportion of Treg in KO mice was significantly higher than that in Wt littermates after ischemia (
Application of L -histidine Reduced Infarct Volume of Wild-Type Mice, But Not Oct3 Knockout Mice
We speculated that targeted disruption of Oct3 would ameliorate ischemic brain damage by modulating the level of histamine and the population of Treg. To test our hypothesis, we next investigated the effects of
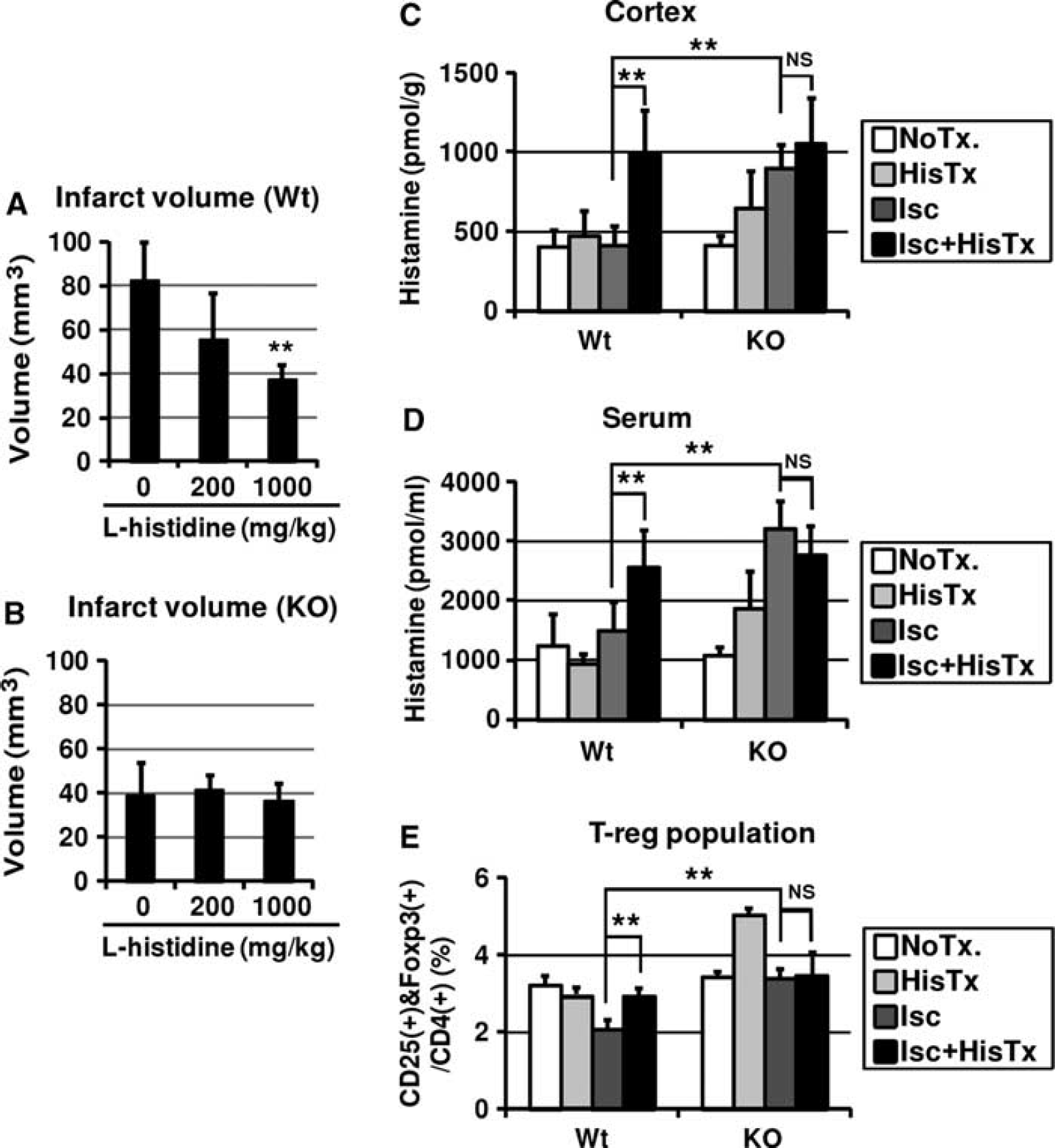
Effects of repeated administration of
Repeated Administration of L -Histidine Modulates Histamine Level and Treg Cell Population after Cerebral Ischemia
Finally, we evaluated histamine level and the proportion of Treg cells at 24 hours after reperfusion in the presence and absence of both treatment with
Without ischemia, repeated treatment with
At 24 hours after reperfusion, histamine level in the I.C. (Figure 7C) and serum (Figure 7D) and Treg cell proportion (Figure 7E) in KO mice was significantly higher than those in Wt littermates.
In addition, at 24 hours after reperfusion, repeated treatment with
Taken together, these results suggest that OCT3 is closely related to histamine clearance after ischemia, and the Treg cell proportion is correlated with the histamine level.
Discussion
In the present study, we showed that targeted disruption of Oct3 ameliorated ischemic brain damage through increases in histamine content and the regulatory T-cell population.
Histaminergic neurons are exclusively located in the tuberomamillary nuclei of the posterior third of the hypothalamus. The histaminergic fibers emanating from the tuberomamillary nucleus project to the cerebral cortex, thalamus, basal ganglia, basal forebrain, and hypothalamus (Haas et al, 2008). Histamine content in the brain has been reported to increase transiently after cerebral ischemia in primates (Subramanian et al, 1981) and rodents (Adachi et al, 1992). We also showed that histamine level increased up to 6 hours after reperfusion in the I.C. and I.S. of Wt littermates. Moreover, histamine level in the I.C. of KO mice was significantly higher than that of Wt littermates during and after ischemia. The released histamine from storage or producing cells into the extracellular space is inactivated to terminate its effects. To remove an excess amount of histamine in the extracellular space, histamine is transported into cells and metabolized into inactive metabolites (Ogasawara et al, 2006). Two distinct transport systems, named uptake-1 and uptake-2, are responsible for the clearance of monoamines including histamine. Among these transport systems, OCT2 and OCT3 are identified as the uptake-2 transporter, and have the ability to transport histamine into cells in a potential-sensitive mode (Grundemann et al, 1999). The OCT3 has been reported to be the molecule responsible for uptake and clearance of histamine in tissues (Ogasawara et al, 2006; Schneider et al, 2005). The OCT2 is expressed exclusively in the kidney, whereas OCT3 is expressed ubiquitously (Burckhardt and Wolff, 2000). The OCT3 might therefore account for uptake and clearance of brain histamine induced by cerebral ischemia. In this study, we showed that repeated treatment with
Many studies have reported that histamine has beneficial roles in cerebral ischemia. Intracerebroventricular administration of histamine and repeated intraperitoneal administrations of
Cerebral ischemia induces rapid inflammatory processes in both the central nervous system and the periphery, followed by long-lasting severe immunodepression. All of these immune reactions contribute to cerebral damage and worse outcomes (Emsley and Hopkins, 2010). Increasing evidence indicates that Treg cells are key cerebroprotective immunomodulators after cerebral ischemia. Severe immunodepression was accompanied by increased CD4+ CD25+ Foxp3+ Treg cells and circulating CD11b+VLA4-negative macrophages (Offner et al, 2006). Depletion of the CD25+ population with anti-CD25 mAb significantly increased brain damage and worsened the functional outcome after cerebral ischemia. Treg cells prevent brain damage by counteracting excessive production of proinflammatory cytokines and by modulating invasion and activation of lymphocytes and microglia in the ischemic brain (Liesz et al, 2009). Depletion of Treg cells augmented postischemic activation of resident and infiltrating inflammatory cells including microglia and T cells, the main sources of cerebral TNF-α and IFN-γ. Development of a cerebral infarct in Treg cell-depleted mice is mediated by TNF-α and IFN-γ (Lambertsen et al, 2004; Liesz et al, 2009). Histamine can influence Th1, Th2, and Treg cell balance and consequently antibody-isotype switching. Histamine can promote Th1-type responses through H1R, whereas it can negatively regulate both Th1- and Th2-type responses through H2R by several different mechanisms (Jutel et al, 2006). Histamine can also positively interfere with peripheral immunological tolerance induced by Treg, through H2R and H4R (Akdis and Akdis, 2011; Morgan et al, 2007). The Treg cells have an essential role in peripheral immunological tolerance. Certain cytokines such as TGF-β, IL-10, and IL-12 may be essential for Treg activation and maintenance of their suppressor functions (Mellor and Munn, 2011). Histamine induces the production of IL-10 by dendritic cells as well as Th2 cells (Akdis and Blaser, 2003). Histamine also enhances the suppressive activity of TGF-β on Th2 cells (Kunzmann et al, 2003).
In this study, we showed that OCT3 was closely related to histamine clearance after ischemia, and the CD4+CD25+Foxp3+ Treg cell proportion was correlated with the histamine level. CD4+CD25+Foxp3+ Treg cells can be classified into two major subgroups; naturally occurring Treg that are constantly produced by the thymus and inducible (or adaptive) Treg that are induced in the periphery on antigenic stimulation of naive T cells under tolerogenic conditions. We cannot confirm which type of CD4+CD25+Foxp3+ Treg cells are actually modulated because there are still no reliable phenotypic or functional markers that make it possible to distinguish between natural and inducible CD4+CD25+Foxp3+ Treg cells. In any case, our results confirm the previous notion that histamine stimulates recruitment of the Treg cell population (Morgan et al, 2007). Since histamine could suppress gamma-delta T cell-mediated cytotoxicity through H2R (Truta-Feles et al, 2010), there is a possibility that gamma-delta T lymphocytes can have pivotal role even in the acute phase of cerebral ischemia (Shichita et al, 2009). But no modulation of gamma-delta T cells population was observed during acute phase of ischemia (data not shown). Our study showed that either targeted disruption of Oct3 or the repeated treatment with
Finally, we showed that Oct3-deficient mice developed smaller infarcts and lower expression of IL-6 and MCP-1 in the I.C. at 24 hours after transient ischemia. IL-6 is a pleiotropic cytokine and MCP-1 a pleiotropic chemokine. They can be key mediators in the inflammatory response to cerebral ischemia. In an experimental stroke model in rodents, many investigators have reported upregulation of IL-6 and MCP-1 after cerebral ischemia (Semple et al, 2010; Suzuki et al, 2009). In patients with acute ischemic stroke, the levels of IL-6 and MCP-1 increase in both serum and cerebrospinal fluid. Serum IL-6 level is associated with greater stroke severity, early neurologic worsening, larger infarct volume, and worse clinical outcome (Suzuki et al, 2009), and serum MCP-1 level is associated with recurrent brain infarction (Kuriyama et al, 2009). It is, therefore, generally accepted that both IL-6 and MCP-1 are predictive markers of the magnitude of the ischemic insult, although the roles of this cytokine and chemokine in cerebral ischemia remain to be elucidated. Since, as far as we know, there is no evidence that histamine directly stimulates the expression of IL-6 or MCP-1, our results suggest that reduced expression of IL-6 and MCP-1 in Oct3-deficient mice may result from reduced ischemic insult. We also showed that there were no significant differences in the levels of IL-12p70 and IL-10 between KO mice and Wt littermates after ischemia, although IL-10 and IL-12 may be essential for Treg activation and maintenance of their suppressor functions (Mellor and Munn, 2011). Our results suggest the possibility that Treg can be activated after ischemia without IL-12p70 and IL-10 signaling. Further investigations are required to clarify the molecular mechanisms of Treg activation after cerebral ischemia.
Footnotes
Acknowledgements
The secretarial assistance of Ms. K Hiraoka is acknowledged.
Disclosure/conflict of interest
The authors declare no conflict of interest.