
Abstract
Inflammation is a highly dynamic and complex adaptive process to preserve and restore tissue homeostasis. Originally viewed as an immune-privileged organ, the central nervous system (CNS) is now recognized to have a constant interplay with the innate and the adaptive immune systems, where resident microglia and infiltrating immune cells from the periphery have important roles. Common diseases of the CNS, such as stroke, multiple sclerosis (MS), and neurodegeneration, elicit a neuroinflammatory response with the goal to limit the extent of the disease and to support repair and regeneration. However, various disease mechanisms lead to neuroinflammation (NI) contributing to the disease process itself. Molecular imaging is the method of choice to try to decipher key aspects of the dynamic interplay of various inducers, sensors, transducers, and effectors of the orchestrated inflammatory response in vivo in animal models and patients. Here, we review the basic principles of NI with emphasis on microglia and common neurologic disease mechanisms, the molecular targets which are being used and explored for imaging, and molecular imaging of NI in frequent neurologic diseases, such as stroke, MS, neurodegeneration, epilepsy, encephalitis, and gliomas.
Keywords
Introduction
Present in all vertebrates, inflammation is a highly dynamic and complex process combining local and systemic reactions of multiple cell types, chemical signals, and signaling pathways. Inflammation is an adaptive response for restoring tissue homeostasis (Medzhitov, 2008) and implies a tight interplay between the tissue and the immune system. It is found in physiological and pathological situations, where it is beneficial when it contributes to support repair and regeneration (e.g., wound healing, scar formation, and suppression of infection) and, on the opposite, detrimental when excessive or persistent inflammation worsens tissue injury (e.g., allergy, chronic infections, and neurodegeneration).
Almost any type of tissue insult activates inflammation; however, the inflammatory response varies with the nature of the insult (mechanical, chemical, infectious, and tumoral) and with the tissue. Originally viewed as an immune-privileged organ, the CNS was progressively recognized to be constantly in direct and dynamic interaction with the innate and the adaptive immune systems. Neuroinflammation (NI) is particular because (1) the CNS has efficient natural protection from mechanical aggressions by the skull and from biological and chemical aggressions by the blood–brain barrier (BBB); (2) the CNS has limited regeneration capacities; and (3) any local impairment of its spatial organization can lead to the definitive loss of a major function. Neuroinflammation is a physiological defense process in which resident microglia and infiltrating immune cells from the periphery have an important role, but it is also involved during brain maturation and neurogenesis. Diseases of the central nervous system (CNS) lead to a neuroinflammatory response, whose trivial goal is to limit the extent of the disease, to clear tissue damage and to support repair and regeneration. However, there is also clear evidence in several cases that NI may, depending on the inducing pathological mechanism, contribute to the disease process itself. More surprisingly, NI seems also to be associated with psychiatric disorders such as major depression (Alexopoulos and Morimoto, 2011) or schizophrenia (Doorduin et al, 2009a).
Given the dynamic nature of NI, the variety of cells and factors involved and the changes in tissue organization induced, exploration methods of practical value for its understanding must take ‘time’ and ‘space’ into account. In many instances, anatomical and functional in-vivo imaging methods are essential for diagnosis and as read-out of effective therapy and have a major impact on patient care. More recently, in-vivo imaging technologies have matured enough to look into the molecular mechanisms and functional consequences of neuroinflammatory processes at various disease stages, and even though we are still struggling through the ‘jungle’ of chemical effectors, receptors, signaling mechanisms, and cellular interactions involved, in-vivo cellular and molecular imaging is increasingly called on to improve our understanding of NI. Conversely, the objective of molecular imaging is not only to decipher further the cellular biochemistry of NI, but also to develop efficient, reliable, and quantitative noninvasive methods capable to guide therapeutic developments that alter neuroinflammatory cascades, prevent tissue damage and support tissue repair processes. At the same time, care must be taken not to overemphasize the clinical importance of NI during the disease process, just because imaging studies provide evidence of its presence, which even though genuine may have little practical importance. Future work should therefore concentrate on the dynamic interplay between NI and the molecular mechanisms inducing cellular damage.
The intention of this article is to review NI imaging strategies which have been used in experimental and clinical applications. Rather than focusing on a single target, imaging method, or disease mechanism, we describe and discuss the current status and possible future developments of (1) molecular targets for NI, (2) imaging methods and technologies, in (3) a variety of neurologic diseases. This review shall complement other reviews that guide the reader through the advantages and disadvantages of a particular imaging modality (e.g., Stoll and Bendszus, 2010 and Wunder et al, 2009). Instead, we focus on molecular mechanisms and possible targets for imaging in various neurologic diseases, assuming that the reader is familiar with the basic principles of the major imaging technologies, such as optical, radionuclide (positron-emission tomography (PET), and single-photon emission computed tomography (SPECT)), and magnetic resonance imaging (MRI), as their different strengths and weaknesses have already been extensively commented upon, i.e., a high sensitivity and high spatial resolution for optical; high sensitivity and possibility for true signal quantification for radionuclide imaging; and high spatial resolution and physiological and biochemical imaging for MRI.
The review is divided into a first part, in which the basic principles of NI are grossly summarized with special emphasis on microglia. In the second part, several molecular targets among the complex interplay between inducers, transducers, and effectors of NI are selected and discussed, in particular those which are candidates to or are already used as targets for noninvasive molecular imaging. The third part deals with clinical and experimental molecular imaging of NI in the most frequent neurologic diseases (stroke, multiple sclerosis (MS), neurodegenerative diseases (ND), epilepsy, encephalitis, and gliomas).
Basic principles of neuroinflammation
The term ‘inflammation’ refers to a generic multicellular process, characterized by (1) changes in local vasculature (increased blood flow and vascular permeability), (2) activation of resident immune competent cells, (3) infiltration of mobile cells of the immune system (neutrophils, macrophages, and lymphocytes), and (4) cytokine production (Graeber et al, 2011). Neuroinflammation is the inflammation of the nervous system observed in diseases of the CNS, including stroke, MS, Alzheimer's (AD) and Parkinson's disease (PD), neurotrophic viral infections, neoplasias, head traumas, and even excess ethanol absorption. All these pathologies trigger an immune activation of the brain, which contributes on the one hand to tissue damage, loss of neurons and dysfunction, on the other hand is involved in neuroregeneration and tissue repair (Rivest, 2009). Signals triggering inflammation may vary depending on the cause or inducing factor. Using optical in zebrafish, hydrogen peroxide (H2O2) was recently identified as the very first signal for wound detection and the primer of inflammation (Niethammer et al, 2009). It is not unlikely that H2O2 is also the signal initiating NI in the mammalian brain, although this remains to be demonstrated. Between the myriad of primary and secondary signaling molecules involved at different levels in NI, the role of ATP in triggering the activation of microglia cells is well established and was beautifully shown in live mouse brains by two-photon microscopy (Davalos et al, 2005).
Neuroinflammation is classically associated with CNS infections (e.g., herpes simplex virus type 1 (HSV-1) encephalitis) and autoimmune disease (e.g., MS) as well as with acute (e.g., ischemia) or chronic (e.g., AD) CNS disease processes. The NI occurring in the absence of microorganisms has been termed as sterile inflammation (Chen and Nunez, 2010). In chronic CNS diseases, such as AD and PD, some authors have proposed to replace the term ‘neuroinflammation’ with the term ‘microglial activation’ (Graeber et al, 2011) with respect to the leading role of microglia and the absence of an obvious participation of granulocytes or T cells in these diseases.
Neuroinflammation can be viewed as the immune response of the brain, a symphony played by an orchestra of many cells and molecules and involving an intricate combination of events with varying time courses:
Activation of damage-associated molecular patterns (DAMPs; e.g., high-mobility group box 1 proteins, heat-shock proteins, histones, oxidized lipids, ATP, amyloid-β (Aβ)) by tissue injury, or of pathogen-associated molecular patterns by exogenous pathogens; Activation of Toll-like receptors (TLRs) of various subtypes and of the receptor for advanced glycation end products with subsequent release of adhesion molecules, cytokines, and chemokines directing activation and targeted migration of effector cells (microglia, macrophages, and lymphocytes); Activation and recruitment of microglial cells, the major cell of innate immunity in the brain; Infiltration of macrophages and T lymphocytes of various subtypes (Th1, Th2, Th17, and regulatory T cells); Alteration of the BBB that is designed to prevent entry of T cells and neurotoxic compounds at the neurovascular unit; Activation and recruitment of astrocytes that tend to control T-cell infiltration into the CNS by inducing apoptosis; Release or expression of a variety of signaling factors, including the neuron-derived neuropeptides, membrane proteins (e.g., fractalkine (FKN), cannabinoid receptors, and major histocompatibility complex molecules), semaphorins and lectins that all take part in the regulation of the neuroinflammatory process (reviewed by Amor et al, 2010; Macrez et al, 2011; Neumann et al, 2009; and Tian et al, 2009; Figure 1).
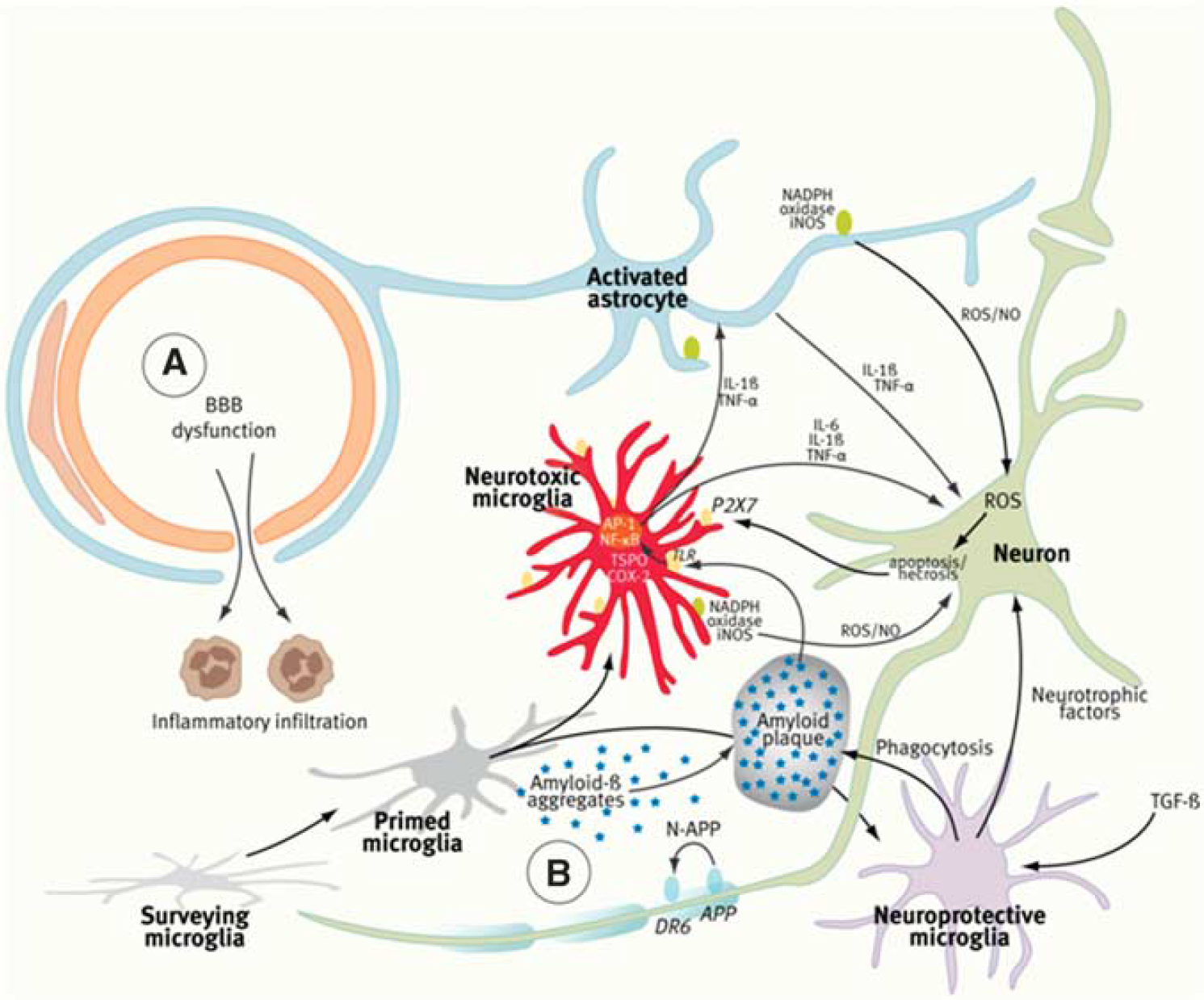
Components of the neurovascular unit (NVU), the ‘Vicious Cycle’ of neuroinflammation, and the ‘Ying and Yang’ of microglia. In ischemic stroke and multiple sclerosis (MS) (
The main actors of the innate (nonspecific) immune response of the CNS are the microglial cells that constitute ∼10% of the entire cell population of the brain. Microglia represent the brain's resident macrophages involved in the removal of cell debris after ischemia or myelin damage, in the limitation of neurotrophic viral infections, as well as in neurorepair processes. Microglia are stimulated by and contribute to a variety of brain diseases (Perry et al, 2010) and, depending on the injury and microenvironmental conditions, may aggravate injury and cause neurodegeneration, or conversely may mediate protective mechanisms promoting tissue repair and regeneration, in opposing roles that have been termed as ‘the Yin and Yang’ of microglia (Czeh et al, 2011). Microglia are essential for normal brain maturation and involved in various developmental processes such as apoptosis, axonal growth and guidance, regulation of embryonic cortical precursor cell development, neuronal differentiation, astrocyte proliferation, and angiogenesis (Czeh et al, 2011). In the adult brain under normal physiological conditions, microglia exhibit a noninflammatory phenotype often described as quiescent or resting. However, imaging studies of fluorescently tagged microglia have shown that, as ‘cops on the beat’ (Raivich, 2005), microglial cells exert active and permanent surveillance of their local environment and produce anti-inflammatory and neurotrophic factors (Davalos et al, 2005; Nimmerjahn et al, 2005). After activation by various stimuli (tissue damage, pathogen invasion, and protein aggregates), microglia switch to an activated phenotype and promote an inflammatory response to serve pathogen clearance and tissue repair (Davalos et al, 2005). Reactive microglial cells derived from resident microglia have morphological features similar to infiltrated macrophages derived from bone marrow (BMD). As the main immune cells of the CNS, microglial cells come in two major subtypes: parenchymal versus bone marrow derived microglia cells with, respectively, low and high major histocompatibility complex class II expression, corresponding respectively to a poor or high antigen-presenting function (Turrin and Rivest, 2006). A recent study suggests that bone marrow derived cells do not enter the healthy CNS in the adult animal and that microglia and blood-derived monocytes have distinct embryonic origins: microglia seed the CNS early during embryogenic development while, thereafter, there is no significant contribution from adult hematopoietic stem cells to the resident pool of microglia in the normal brain (Ginhoux et al, 2010).
The involvement of microglia in developmental processes (apoptosis, axonal growth, and guidance) may be related both to neurorepair mechanisms, especially in the context of their ability to secrete various growth factors, such as brain-derived neurotrophic factor, basic fibroblast growth factor, and insulin-like growth factor (Czeh et al, 2011) and to inflammation-related neurotoxicity. Indeed, several inducers, sensors, transducers, and effectors seem to contribute both to the orchestrated inflammatory response and to the microglia-mediated neurotoxicity (Rivest, 2009) and expression of various surface receptors, including those for complement, cytokines, chemokines, major histocompatibility complex II, and others, trigger or amplify the innate immune response. The simplest, though schematic, manner to describe microglial activation is to use the similarity with the two macrophage types described in peripheral inflammation. Depending on the mode of activation and environmental factors such as age, surveying microglia may come into two phenotypes: the proinflammatory ‘M1’ phenotype is activated by lipopolysaccharides and interferon-γ and corresponds to the ‘classical’ pathway of macrophage activation, while the anti-inflammatory M2 phenotype is activated by interleukin (IL)-4 and IL-13 through the ‘alternative’ pathway of macrophage activation. M1-type microglia produce tumor necrosis factor-α, IL-1β, IL-6, nitric oxide, superoxide, hydrogen peroxide, and matrix metalloproteinases. The M1 microglia phenotype has a central role in host defense against pathogens and tumor cells but also triggers damage to healthy neurons (Czeh et al, 2011). In contrast, the anti-inflammatory M2 microglia phenotype expresses IL-10 and arginase-1 and promotes tissue remodeling/repair and angiogenesis (Czeh et al, 2011). Such a classification in two phenotypes is probably too schematic as the diversity of microglial phenotypes that can be observed in vivo is obviously larger (Olah et al, 2011). Among open questions are (1) the nosology of intermediate states between ‘surveying’ and ‘activated’ microglia and the existence of ‘primed’ microglia (Ferrari and Tarelli, 2011); (2) the molecular mechanisms responsible for the switch between microglial phenotypes (Parkhurst and Gan, 2010); and (3) the control exerted by other cells on microglia activation or deactivation. Concerning this latter point, astrocytes have been claimed to modulate the levels of microglial production and release of reactive oxygen species (Min et al, 2006; Shih et al, 2006). The role of microglia in neurogenesis and aging (reviewed in detail by Gemma et al, 2010) pin-pointed out important interactions between neuron-derived FKN (also known as CX3CL1) and microglial expression of its receptor CX3C chemokine receptor 1 (also known as FKN receptor) for the control of microglial function. Disruption of this dialog triggers the induction of an M1 microglia phenotype and a subsequent increase in the expression of IL-1β and TNF-α. Since aging is associated with a decrease in FKN expression, FKN/CX3C chemokine receptor 1 signaling is disrupted in aged individuals and this leads to an increase in microglial activation and overexpression of proinflammatory cytokines (Cardona et al, 2006; Gemma et al, 2010).
The rational design of molecular imaging targets would greatly benefit from a more thorough understanding of the microglial phenotype–function relationships as discussed in Olah et al (2011). It seems likely that future studies will help to better define the different microglia populations, based both on the nature of the stimulus that provokes the initial insult and on subsequent secondary events that influence the microglial phenotype, and that this will help to understand the influence of microglial phenotypes on the outcome of CNS injuries and pathologies (Perry et al, 2010). Given our presently limited knowledge of microglial dynamics, it may appear simpler at this stage to distinguish between the activated microglia found in acute inflammation (mechanical brain damage, transient infections, and stroke) and the chronically activated microglia found in neurodegenerative disorders or chronic infections (Parkhurst and Gan, 2010). However, practical biomarkers have yet to be validated for an irrefutable recognition of these two microglial states.
Molecular targets for imaging neuroinflammation
All molecular imaging techniques, including two-/multiphoton and fluorescence microscopy, PET, SPECT, and MRI are used experimentally and clinically to decipher molecular alterations of neurologic disorders. Here we place the emphasis on the molecular imaging targets directly or indirectly connected to NI.
Damage-Associated Molecular Patterns/Pathogen-Associated Molecular Patterns
A major recent breakthrough for in-vivo molecular imaging of disease-specific molecular alterations has been the direct visualization of amyloid-β (the DAMP of AD) using thioflavine and multiphoton microscopy (Bacskai et al, 2001). Direct assessment of the amyloid plaque burden and the effect of immunomodulatory therapy helped the development of radiolabeled thioflavine derivatives (e.g., Pittsburg compound B; [11C]PIB) for imaging Aβ in humans (Klunk et al, 2004). Combined imaging of Aβ and microglial activation is given special attention in patients with presymptomatic AD and mild cognitive impairment (MCI), in the hope to answer the question whether Aβ (as DAMP) or activated microglia (as the main parameter of NI) is the disease-driving mechanism leading to ND (Okello et al, 2009).
Another DAMP linked to NI is the induction of heat-shock proteins that can be assessed in vivo either by reporter systems using heat-shock protein-sensitive promoter systems (Deckers et al, 2009; Doubrovin et al, 2011) or by directly targeting probes such as [18F]fluoromethyldeoxyspergualin (Ghosh et al, 2011).
Toll-Like Receptors, Receptor for Advanced Glycation End Products, Adhesion Molecules, Cytokines, and Chemokines
The past years have witnessed technical advances for investigating the functional associations of TLRs and other pattern recognition receptors using noninvasive fluorescence imaging methods in living cells (reviewed by Triantafilou and Triantafilou, 2012). Independently from imaging, TLRs (e.g., TLR7,8) and cytokines and chemokines (IL-1β and IL-6) are also being used as biomarkers for tissue outcome after stroke (Brea et al, 2011). New TLR-directed therapies (e.g., anti-TLR antibodies) are now evaluated in conjunction with imaging methods that assess tissue outcome (e.g., μMRI after experimental myocardial infarction; Arslan et al, 2010), or with atomic force microscopy imaging to investigate the fibrillar morphology of proinflammatory Aβ(1 to 42) species (Udan et al, 2008).
Microglia Cells
Microglial responsiveness to injury places these cells as diagnostic markers of disease onset or progression (Perry et al, 2010). As depicted in Figure 1, various cell surface and mitochondrial receptors expressed in microglia cells are involved in the regulation and function of microglia, and some of these receptors have been used for the development of ligands for imaging.
Translocator protein
Upregulation of the translocator protein 18 kDa (TSPO, formerly called peripheral benzodiazepine receptor or PBR) is a hallmark of activated microglia. Translocator protein is a protein of the outer mitochondrial membrane associated with a voltage-dependent anion channel and a nucleoside transporter. It is primarily involved in the transport of cholesterol into mitochondria, which is the rate-limiting step in the synthesis of steroids and neurosteroids (Rupprecht et al, 2010). In the CNS, TSPO is highly expressed in activated microglia, in the choroid plexus and to a lesser extent in reactive astrocytes, in neurons of the olfactory bulb, and in neuroblastoma and glioblastoma cell lines (Rupprecht et al, 2010), but its expression is globally low in the normal brain. Translocator protein is a sensitive biomarker for microglial activation and reactive gliosis (Chauveau et al, 2008), and binding to TSPO of radiolabeled ligands can be visualized by PET and SPECT by a variety of ‘old’ ([11C](R)-PK11195) and ‘novel’ radioligands, such as [11C]DAA1106, [18F]FE-DAA1106, [11C]DPA-713, [18F]DPA-714, [18F]PBR28, [18F]PBR111, [11C]SSR18075, [11C]CLINME, [123I]CLINDE, and [11C]vinpocetine (Arlicot et al, 2008; Banati et al, 1997; Chauveau et al, 2008, 2011, 2009; Ciarmiello, 2011; Dolle et al, 2009; Imaizumi et al, 2007; James et al, 2008; Kiferle et al, 2011; Van Camp et al, 2010; Winkeler et al, 2010). Comparison of ‘novel’ (e.g., [11C]vinpocetine, [18F]DPA-714, [18F]PBR111, and [11C]SSR180575) with ‘old’ ([11C](R)-PK11195) TSPO ligands in rodent models and patients with NI revealed improved bioavailability, decreased nonspecific uptake, and higher specific binding of the novel compounds (Chauveau et al, 2011, 2009; Van Camp et al, 2010; Vas et al, 2008). However, different binding affinity patterns have been identified in humans and should be taken into account when interpreting the imaging findings (Owen et al, 2011). Moreover, it has to be taken into account that various TSPO ligands might exert different biological effects on microglial activation with respect to microglia proliferation and phagocytosis (Choi et al, 2011; Veiga et al, 2007).
Further microglial targets
Further microglial targets currently being explored for imaging include the P2X7 receptor (Monif et al, 2009; Yiangou et al, 2006), the cannabinoid CB2 receptor (Evens et al, 2009; Horti et al, 2010; Turkman et al, 2011; Vandeputte et al, 2011), the cyclooxygenase-1 and -2 enzyme (de Vries et al, 2008; Shukuri et al, 2011), and matrix metalloproteinases (Iwama et al, 2011; Pinas et al, 2009; Wagner et al, 2007). The CB2 receptors can be targeted by both radiolabeled and paramagnetic imaging probes (te Boekhorst et al, 2010).
There is obviously a discrepancy between the multitude and diversity of cellular and molecular players in NI on the one side and the relatively small number of currently established molecular tracers on the other side. The TSPO (PBR) imaging is a preferred method of many experimental and clinical studies and to develop more specific TSPO tracers may be helpful. Nevertheless, in a way the progress made for example by DPA-714 as compared with PK11195 obscures the fact that tracers targeting other mechanisms are still very rare to date.
Infiltrating Neutrophils, Macrophages, and T Lymphocytes
The recruitment of circulating leukocytes to the site of injury is induced by upregulation of endothelial adhesion molecules (P/E-selectin, intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1); Man et al, 2007). Various molecular contrast media to target selectin, ICAM, and VCAM expression have been developed. They are based, for example, on (1) 125I-labeled gold nanorods (GdNRs) or 64Cu-labeled nanoparticles conjugated with anti-ICAM-1 antibody (Rossin et al, 2008; Shao et al, 2011); (2) GdNRs for photoacoustic detection (Ha et al, 2011); (3) anti-CD34 microbubbles as targeted ultrasound contrast agents (Liu et al, 2011); and anti-ICAM-targeted echogenic immunoliposomes (Kiessling et al, 2009; Kim et al, 2010a); (4) anti-VCAM antibody conjugated microparticles of iron oxide (Hoyte et al, 2010; Leung, 2011; McAteer et al, 2007); or (5) double-conjugated fluorescent microspheres in conjunction with laser scanning ophthalmoscopy (Sun et al, 2010). Determinations by enzyme-linked immunosorbent assay of soluble E-selectin, ICAM-1, VCAM-1, together with measurements of matrix metalloproteinase-9, tissue inhibitor of metalloproteinase 1, plasma TNF-α and IL-6, are also used as biomarkers for NI in association with conventional MRI to predict tissue injury and stroke severity in early ischemia (Bogoslovsky et al, 2011).
Studies of the kinetics of T-cell motility and transmigration by direct cell imaging by dynamic two-photon microscopy (Soriano et al, 2011; Svensson et al, 2010) have revealed a sequential involvement of endothelial ICAM-1 and VCAM-1 in mediating shear-resistant T-cell arrest, followed by endothelial ICAM-1 and ICAM-2 mediating T-cell crawling to sites permissive for diapedesis across BBB endothelium (Steiner et al, 2010). Sorokin has pointed out the importance of extracellular matrix components influencing immune cell infiltration through the BBB (Sorokin, 2010).
Unspecific labeling of leukocytes or macrophages is performed ex vivo by incubation of white-blood cells with 111In- or 99mTc-labeled compounds for SPECT or [18F]FDG for PET imaging (reviewed by Wunder et al, 2009), or in vivo using MRI nanosized/ultrasmall agents such as iron-oxide nanoparticles (Stuber et al, 2007), liposomes encapsulating monodisperse single core superparamagnetic iron-oxide particles (Soenen et al, 2010) or paramagnetic lanthanide-based agents (Castelli et al, 2009; reviewed by Stoll and Bendszus, 2010). The extent of macrophage labeling in relation to clearance by the reticulo-endothelial system depends on particle size, coating, and route of in-vivo delivery. The drawbacks of unspecific cell labeling methods are the possible leakage of the label from the cells and unspecific accumulation in the brain due to a disrupted BBB (Stoll and Bendszus, 2010; Wunder et al, 2009). These issues can be overcome by using genetic engineering of cells expressing artificial marker genes, in which specific accumulation and trapping of the label occurs selectively (Costa et al, 2001; Waerzeggers et al, 2008, 2009). Another approach is to conjugate a fluorescent probe, such as Cy5.5, by the HIV-TAT system to T lymphocytes, which are then adoptively transferred into experimental animals (Berger et al, 2007). Finally, [18F]FDG μPET/computed tomography can follow experimental autoimmune encephalitis (EAE) coinciding with increased glucose consumption, presumably by infiltrating immune cells in the spinal cord (Radu et al, 2007).
Alterations of and Within the Blood–Brain Barrier
Changes in the vascular permeability and the BBB can be assessed by gadolinium-diethylenetriaminepentaacetic acid (Gd-DTPA) and MRI or by 99mTc-pertechnetate (99mTeO4) or 99mTe-DTPA and SPECT in vivo (reviewed by Wunder et al, 2009). Magnetic resonance imaging with Gd-DTPA is used on a routine clinical basis to assess the activity and extent of BBB disruption after ischemic stroke, and during the disease course of encephalitis, MS, and gliomas, and it may serve as noninvasive imaging biomarker to assess the efficiency of treatment with corticosteroids.
Astrocytes and Monoamine Oxidase Type B
Monoamine oxidase type B is an enzyme located at the outer mitochondrial membrane primarily in astrocytes and serotoninergic neurons. Monoamine oxidase type B catalyzes the deamination of monoamines, thereby influencing neurotransmitter concentrations, and its activity detected with [11C]-
Imaging neuroinflammation in neurologic diseases
This part summarizes experimental and clinical neuroimaging studies of NI during the course of major neurologic diseases, i.e., stroke, MS, AD, PD, as well as in amyotrophic lateral sclerosis (ALS), epilepsy, encephalitis, and gliomas.
Stroke: Stroke is the most common neurologic disorder, the third leading cause of death in the United States and the leading cause of serious, long-term disability. In 80% of stroke cases, occlusion of a cerebral vessel leads to cerebral ischemia and only immediate establishment of reperfusion can substantially improve functional outcome (NIND, 1995; Sobesky et al, 2007). Occlusion of a cerebral artery induces ischemic infarct of the corresponding cerebral territory, with a cascade of metabolic and inflammatory consequences that extend into the peri-infarct zone (penumbra). Activation of microglia and astrocytes and recruitment of leukocytes contribute in part to the cell damage (Dirnagl et al, 1999; Macrez et al, 2011). A retrograde degeneration of neurons as well as anterograde Wallerian degeneration of axons occurs in cerebral areas distant from the focal ischemia-induced inflammatory area. This is accompanied by the activation of microglia along the degenerating fiber tracts, which may persist for several years (Perry et al, 2010).
As reviewed recently (Allan et al, 2005; Macrez et al, 2011; Rivest, 2009; Wang et al, 2007), DAMP-induced synthesis of proinflammatory cytokines (TNF-α, IL-1β, and IL-6), microglial activation and leukocyte infiltration have a major role in the pathogenic events after cerebral ischemia: cellular reactions determine the extent of ischemia-induced tissue damage but are also necessary for tissue repair and regeneration in the advanced stage. Tumor necrosis factor-α and IL-1β are rapidly produced in response to ischemic injury and directly damage neurons, endothelial and glial cells, and recruit circulating leukocytes to the site of injury by inducing upregulation of endothelial adhesion molecules (e.g., P/E-selectin, ICAM-1, and VCAM-1). Cellular infiltration into the brain, at first of neutrophils and later of macrophages and lymphocytes (Gelderblom et al, 2009) contributes to postischemic tissue damage through activation of nitric oxide synthase and cyclooxygenase-2 pathways. While gross lymphocyte infiltration is associated with deleterious effects in stroke, regulatory T cells seem to have a cerebroprotective function by counteracting TNF-α and IFN-γ production (Liesz et al, 2009; Lo, 2009). Microglial activation triggers phagocytosis of necrotic cells and secretion of neurotrophic factors (brain-derived neurotrophic factor and insulin-like growth factor; Madinier et al, 2009). To what extent bone marrow derived macrophages precursor cells from the blood can infiltrate the brain and contribute to tissue damage or repair is still a matter of debate and further research (Rivest, 2009; Shichita et al, 2009; Yong and Rivest, 2009). The exact mechanisms that drive macrophage recruitment to the CNS need to be further characterized to explore the possibility to target these cells for therapy. Alternatively, therapeutic strategies that interfere with DAMP-receptor pathways (TLRs) might be a promising approach to restrict the inflammation processes (Macrez et al, 2011).
As many of the mechanisms and pathways described in stroke pathogenesis may be either detrimental or beneficial depending on the temporal disease dynamics, in-vivo imaging of the temporal profile of key events (e.g., TLR activation, microglial activation, and lymphocyte infiltration) appears an important prerequisite for the development and implementation of new therapeutic paradigms (Fagan et al, 2010). Overall, a better understanding of the specific roles of inflammatory cells is needed, in particular with respect to the kinetics of their recruitment and their relative contribution to the evolution of stroke (Macrez et al, 2011).
This is particularly important for a more precise evaluation of the current therapeutic attempts to modify or alter the poststroke inflammation to improve the clinical outcome. Whereas imaging in the acute phase of stroke by computed tomography, MRI, and PET aims to delineate hypoperfused ‘tissue at risk’ surrounding the infarcted territory (penumbra, mismatch), which is still viable and may be rescued by therapy (Zaro-Weber et al, 2010), imaging at later stages after ischemic stroke targets mostly ischemia-induced NI. The MRI-based approaches use systemically administered ultrasmall superparamagnetic iron oxide (USPIO) to track macrophage infiltration into the ischemic area (Stoll and Bendszus, 2010). This approach is however complicated by passive diffusion of free USPIO through a defective BBB (Desestret et al, 2009). A myeloperoxidase-activatable paramagnetic sensor has been developed and used experimentally to noninvasively assess leukocyte- and microglia-related myeloperoxidase activity within the infarct area (Breckwoldt et al, 2008). Attempts for combined USPIO based with perfusion-weighted and diffusion-weighted MR imaging in the clinical application are ongoing. The first studies have revealed conflicting results with regards to the high variability of extent and distribution of USPIO enhancement in relation to infarct size and tissue outcome (Nighoghossian et al, 2007; Stoll and Bendszus, 2010).
Activated microglia have been the target of a number of experimental and clinical studies focusing on studying NI after focal cerebral ischemia. In experimental models of stroke, activated microglia revealed by [11C](R)-PK11195 and μPET are located mainly in the core and in the margin of focal cerebral ischemia between 4 and 7 days after transient ischemia (Rojas et al, 2007; Schroeter et al, 2009). Moreover, activated astrocytes were observed in a rim surrounding the epicenter of the lesion (Rojas et al, 2007). Using [18F]DPA-714 and μPET imaging in a rat model of transient ischemia, the time course of TSPO expression reflecting microglia and macrophage activation within the first 3 weeks after unilateral middle cerebral artery occlusion was described (Figure 2). Expression of TSPO in microglia and macrophages increased until 7 to 11 days after stroke and decreased later. In contrast, the centripetal migration of astrocytes toward the lesion reflecting formation of a scar was correlated with TSPO expression in astrocytes at later times (Martin et al, 2010). Interestingly, minocycline treatment of middle cerebral artery occlusion rats was able to reduce TSPO expression as shown by [18F]DPA-714 PET imaging, but this reduction did not correlate with a reduction in the size of the infarct (Martin et al, 2011). Interestingly, in a model of migraine in rats, [11C](R)-PK11195-PET showed TSPO activation after generation of unilateral cortical spreading depression, indicating that microglia cells are activated also in response to a nociceptive stimulus (Cui et al, 2009).
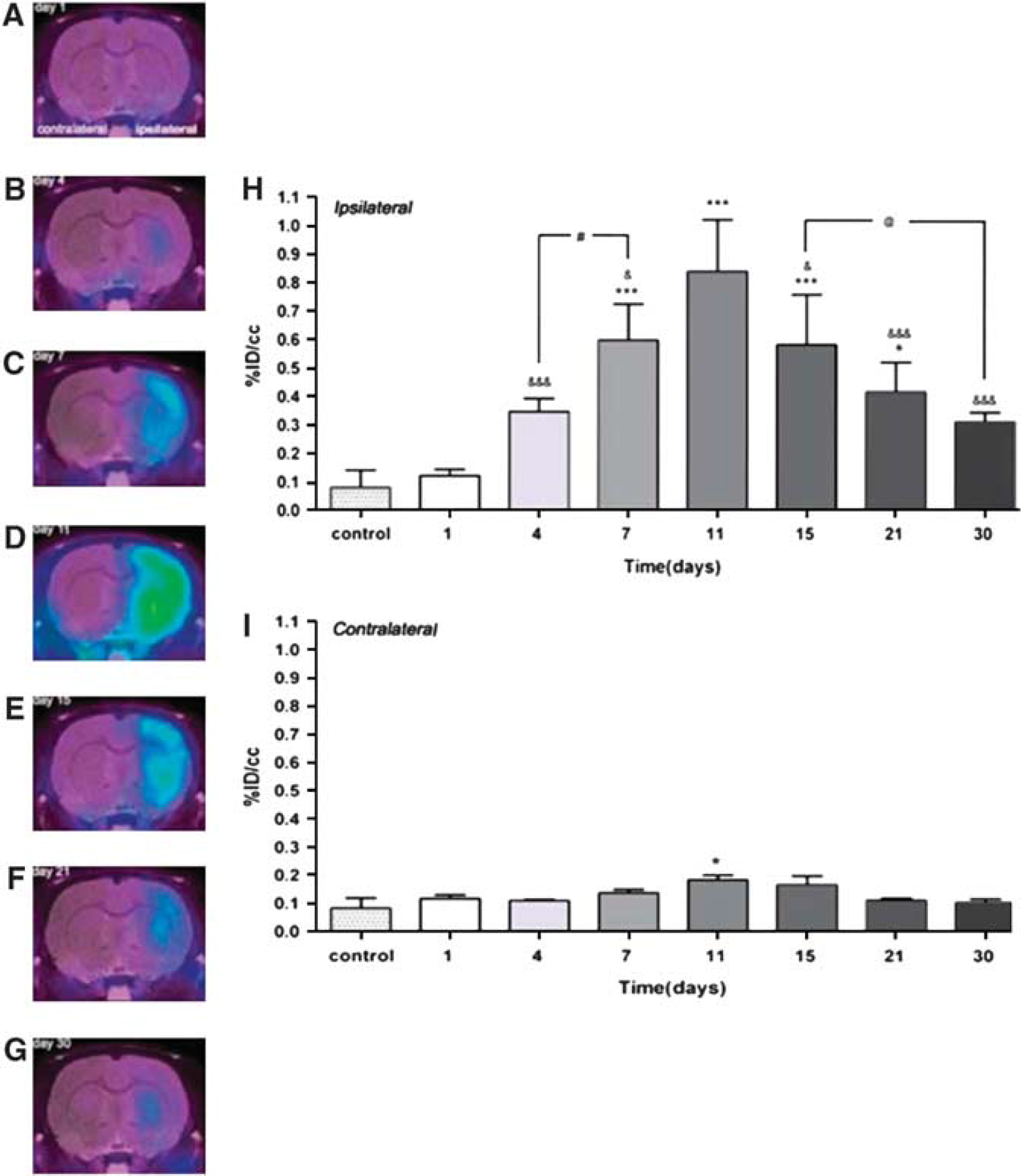
Time course of microglia activation after experimental ischemia as determined by [18F]DPA-714 and μPET. Panels (
Expression of TSPO has been imaged using [11C](R)-PK11195 and PET in relatively small groups of patients with ischemic stroke (Pappata et al, 2000, n=7; Gerhard et al, 2005, n=6; Price et al, 2006, n=4; Radlinska et al, 2009, n=21), or with major risk factors of stroke (atherosclerosis, hyperlipidemia, and obesity; Drake et al, 2011, n=4). Between 3 and 150 days after stroke onset, microglia activation reflected by TSPO ligand uptake can be observed at the primary lesion site, in peri-infarct regions as well as in remote locations, and even in the contralateral hemisphere indicative of pathological changes after Wallerian degeneration (Gerhard et al, 2005; Radlinska et al, 2009). Based on these results, it has been suggested that therapeutic targeting of NI may be extended to late time windows (Price et al, 2006). The consequences of subcortical stroke lesions with or without affection of the pyramidal tract were compared in an elegant study by Radlinska et al, (2009) combining [11C]-(R)-PK11195-PET with diffusion tensor imaging. A remote activation of microglia was found only in patients in whom the pyramidal tract was affected, and this activation was anterograde to the lesion 2 weeks after stroke. Longer follow-up times showed that local microglia activity around the infarct area decreased over time, but that remote microglial activation along affected pyramidal tracts persisted at 6 months (Figure 3; Thiel et al, 2010). The anterograde microglia activity in the brain stem was linearly related to the extent of pyramidal tract damage in the early and chronic phase after stroke. Interestingly, different microglia activation patterns locally and at distant areas were able to predict outcome, i.e., the remote inflammatory activity was positively related to outcome, indicating a possible neuroprotective role or repair function of microglia along the tract portions undergoing Wallerian degeneration (Thiel et al, 2010). Finally, brain inflammation was found in patients with a constellation of chronic systemic inflammatory conditions, even in the absence of any cerebrovascular event, indicating that systemic inflammation can drive brain inflammation before stroke in a so-called ‘primed’ inflammatory environment (Drake et al, 2011).
Location and extent of microglia activation after focal subcortical ischemia as determined by [11C](R)-PK11195 and HRRT-PET in conjunction with diffusion tensor imaging (DTI) MRI. Various microglia activation patterns can be observed after focal cerebral ischemia in humans depending on the affection of the pyramidal tract and the primary lesion size. Acutely activated microglia at the infarct site that decreases after 6 months (
Multiple sclerosis: Multiple sclerosis is the most common inflammatory demyelinating disease of the CNS with a prevalence ranging between 20 and 150 cases for 100,000 inhabitants. It is pathologically characterized by inflammation, demyelination, gliosis, and axonal injury. T cells and macrophages react with myelin antigens and initiate an immune response (secretion of proinflammatory cytokines, release of toxins, and activation of microglia) that leads to demyelination and axonal damage.
As has been nicely summarized by Ajami et al (2011) and reviewed by Kiferle et al (2011), EAE is a well-studied mouse model of the human disease characterized by extensive infiltration of the CNS by inflammatory cells. Initiation of EAE involves the activation of myelin-specific Th1 or Th17 cells, which in turn trigger the expansion of resident microglia and the recruitment of blood-borne myelo-monocytic cells. The fundamental mechanisms leading to the distinct stages of relapsing and remitting disease and to the associated physical impairment remain controversial (Ajami et al, 2011). Using a combination of parabiosis and myeloablation to replace circulating progenitors without affecting CNS-resident microglia, a strong correlation was found between monocyte infiltration and progression to the paralytic stage of EAE, which can be blocked by the inhibition of chemokine receptor-dependent recruitment of monocytes to the CNS (Ajami et al, 2011). These results point to the essential and disease-driving function of infiltrating cells in the pathogenesis of EAE and MS (Ajami et al, 2011). During early disease, CD4-positive T cells and endogenous microglia activation are responsible for disease initiation before the appearance of functional impairment. It is only the appearance of infiltrating monocytes that correlates with substantial disability, and impairing the chemokine (C-C motif) receptor 2-dependent recruitment of these cells prevents progression from very mild to severe disease. The recruitment and activation of macrophages and microglia have an important role in the removal of damaged tissue and in the facilitation of neural repair but, despite their crucial role in host defense, overactivated microglia induce an excess production of cytotoxic factors, which enhance and amplify the neuronal damage (Figure 1; Kiferle et al, 2011). Thus, in EAE, the infiltration of monocytes appears to represent a pathogenic overreaction of the innate immune system (Ajami et al, 2011). Therefore, imaging the passage of immune cells across the BBB would allow the direct assessment of the effects of novel therapeutics aiming to block the transmigration of immune cells into the CNS for treating CNS-directed autoimmune diseases (Ajami et al, 2011; Luster et al, 2005).
Various MRI-based techniques can assess disease activity and effect of therapy in patients with MS. Conventional T1-weighted Gd-enhanced MRI addresses acute disease activity and T2-weighted MRI quantifies overall tissue alterations depicted as hyperintense lesions (NI and neuroaxonal damage) (Bakshi et al, 2008; Hayton et al, 2012). Other image-based primary outcomes in phase II trials of neuroprotective and reparative strategies in MS are (1) changes in whole-brain volume to gauge general cerebral atrophy as measure of neuroaxonal loss; (2) T1 hypointensity and magnetization transfer ratio to monitor the evolution of lesion damage; and (3) optical coherence tomography findings to evaluate the anterior visual pathway (Barkhof et al, 2009).
In addition to conventional MR sequences used in improved model systems (Tourdias et al, 2011), molecular NI-targeted imaging technologies have used USPIO particles to image macrophage and T-cell infiltration in EAE models of MS (Chin et al, 2009; Stoll and Bendszus, 2010). The myeloperoxidase-activatable paramagnetic sensor mentioned above has been used to follow T1-weighted signal increase in areas of active lesions with myeloperoxidase expression (Chen et al, 2008). More and smaller lesions are detected by this approach than with conventional T1- and T2-weighted MRI. Whether these novel molecular imaging approaches will translate into clinical applications remains to be demonstrated. A first attempt in that direction was reported by Vellinga et al (2008) who showed, in patients with active MS, that USPIO-enhanced MRI may reveal subtle inflammatory activity not visible by conventional MRI and unrelated to Gd-DTPA enhancement (Figure 4). Similar results were obtained with improved amphiphilic macrocyclic Gd complexes that allow a more sensitive detection of BBB changes than Gd-DTPA, indicating that complementary information is revealed by both imaging methods (Ladewig et al, 2009; Stoll and Bendszus, 2010).
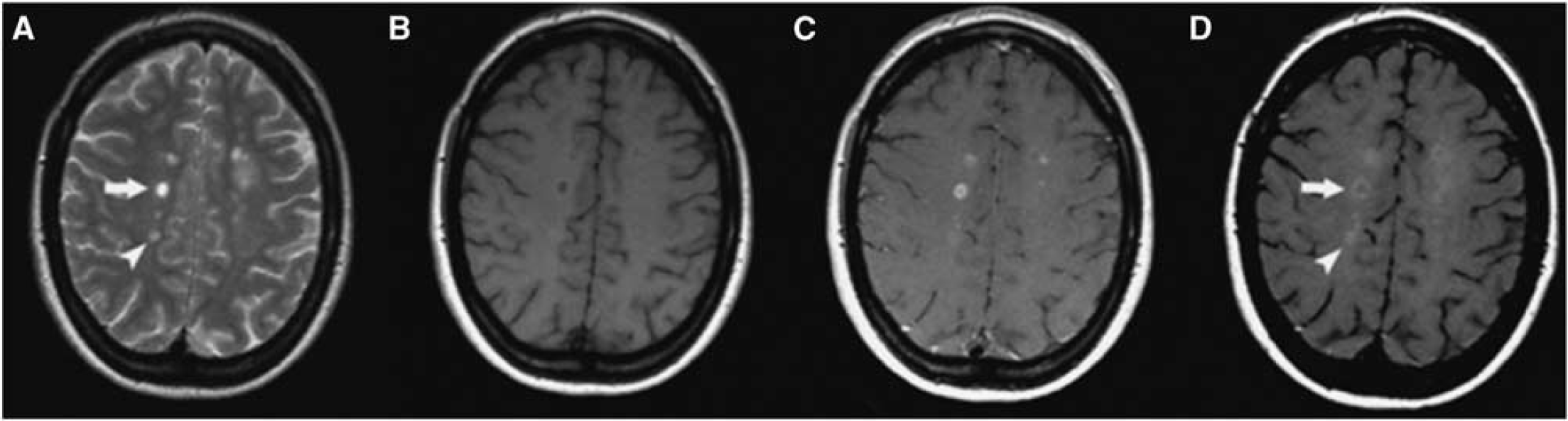
Cross-sectional patterns of lesion enhancement in patients with multiple sclerosis (MS) as detected by magnetic resonance imaging (MRI). (
Experiments based on genetically engineered T cells expressing luciferase have shown that autoantigen-specific CD4+ T cells inhibited inflammation and promoted immunotherapy in the EAE model (Costa et al, 2001). Direct visualization of the local interaction between immune and neuronal cells was shown by two-photon microscopy in living mice subjected to EAE (Siffrin et al, 2010; Figure 5). Direct interaction of Th17 cells specific for myelin/oligodendrocyte glycoprotein with neuronal cells in demyelinating lesions was associated with extensive axonal damage. By combining intravital microscopy with confocal and electron microscopy it could be excellently shown that Th17 cells induce severe, localized, and partially reversible fluctuations in neuronal intracellular Ca2+ concentration as an early sign of neuronal damage, pointing at the key role of the Th17-cell effector phenotype for neuronal dysfunction in chronic inflammation (Siffrin et al, 2010). Moreover, two-photon microscopy provided local quantification of T-cell dynamics, tissue infiltration, and interaction in vivo (Bartholomaus et al, 2009; Kim et al, 2010b; Siffrin et al, 2010). An excellent review on this topic has appeared recently (Kawakami and Flugel, 2010).
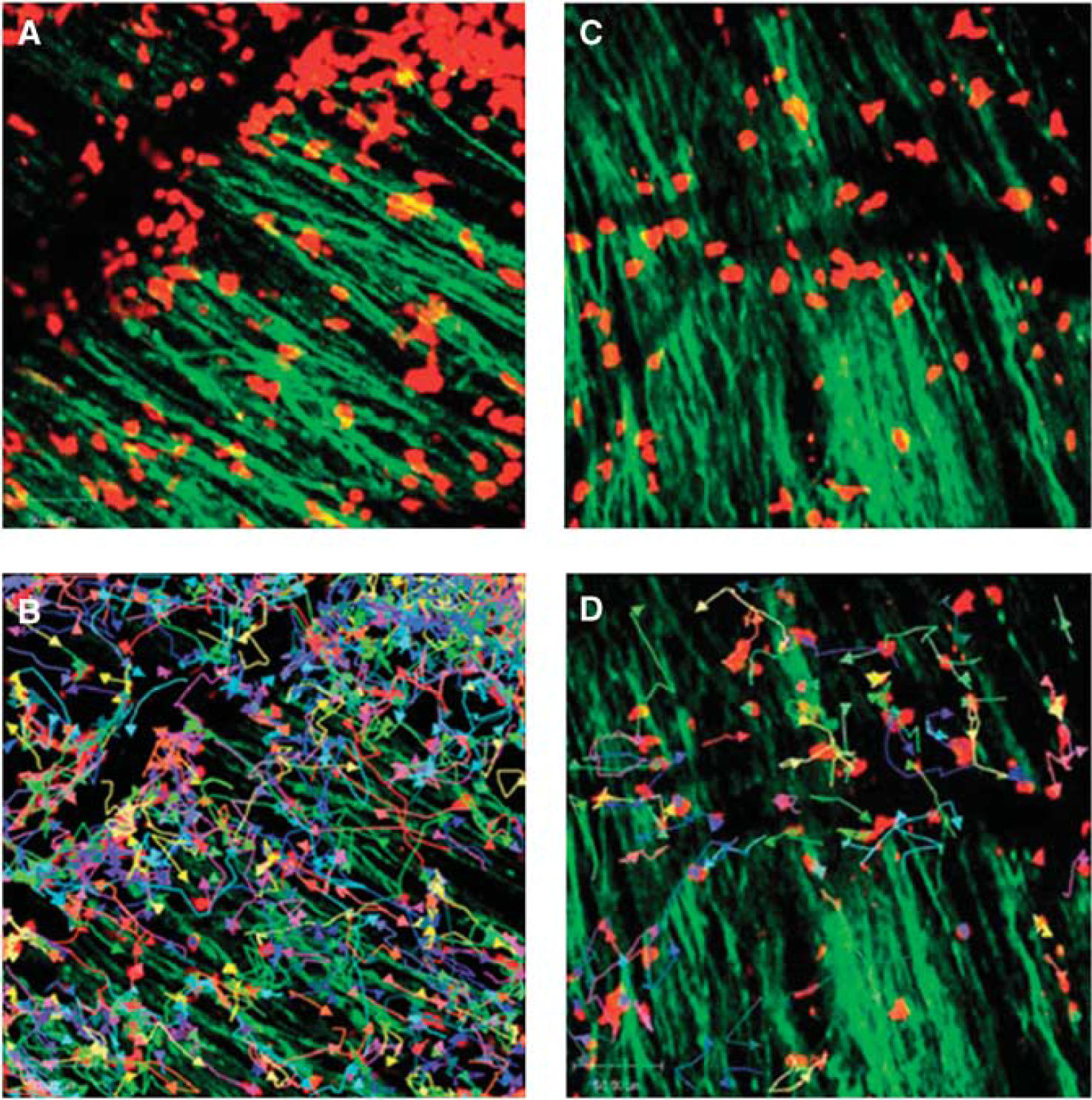
Immune infiltrates in demyelinating lesions as depicted by two-photon laser scanning microscopy (TPLSM) are highly dynamic and show different motility patterns in distinct disease stages. (
Microglial activation in patients with MS has been studied with [11C](R)-PK11195 and PET but so far only in a limited number of patients (Banati et al, 2000, n=12; Debruyne et al, 2003, n=22; Vas et al, 2008, n=4; Versijpt et al, 2005, n=22). Radiotracer binding was increased in areas of acute and relapse-associated inflammation detected by Gd-DTPA enhanced T1-weighted MRI. Interestingly, a significant increase in [3H](R)-PK11195 or [11C](R)-PK11195 binding on activated microglia outside the histopathologically or MRI defined borders of MS plaques was observed in (1) cerebral central gray-matter areas that are not normally reported as sites of pathology in MS (Banati et al, 2000) as well as in (2) normal appearing white matter (Debruyne et al, 2003). This suggests that imaging microglial activation in patients with MS may serve as a complementary biomarker for disease activity staging. Engineered mouse models show that the activation of astroglia and microglia may occur before obvious clinical signs appear in the EAE model of MS (Luo et al, 2008, 2007). A recent study of cuprizone-induced NI in C57BL/6 mice showed positive [123I]-CLINDE accumulation in various brain regions during the phase of demyelination, and a decreased uptake during the phase of remyelination correlating with activated astroglia and microglia cells (Mattner et al, 2011). Similarly, Abourbeh et al (2012) have shown that PET imaging with [18F]DPA-714 can detect the microglial activation in the spinal cord of EAE-induced rats. In summary, visualizing activated microglia in gray matter gives TSPO tracers a promising role in improved understanding of cortical MS pathologies in humans (Kiferle et al, 2011).
Neurodegeneration: Neurodegenerative diseases, such as AD, PD, frontotemporal dementia, Huntington's disease, and ALS, are the most common chronic neurologic disorders and they pose an increasing burden on our aging populations. Alzheimer's disease alone affects at least 34 million people worldwide and its prevalence is expected to triple in the next decades. In AD, histology shows amyloid-β (Aβ) plaques, neurofibrillary tangles, neuronal loss, and atrophy. Symptoms of AD are loss of memory, progressive impairment of cognition, and neuropsychiatric disturbances. In PD, progressive loss of pigmented dopaminergic neurons in the substantia nigra and other brain stem nuclei occurs and leads to dopamine depletion in the striatum, especially in the putamen. Patients with PD develop motor disturbances (bradykinesia, rigidity, tremor, and postural instability), autonomic dysfunction, cognitive impairment, and depression. Amyotrophic lateral sclerosis is the most common motor neuron disease, affects relatively young patients (∼55 years) and has a projected lifetime risk of 1/2,000. Degeneration of motor neurons leads to progressive muscle wasting/weakness, spasticity, and respiratory weakness leading to death usually within 2 to 5 years. Common to AD, PD, and ALS, (1) normal life expectancy is reduced; (2) impairment of cognitive and motor functions is devastating at the personal and family level and has major impact on the health-care system; (3) only symptomatic (not curative) treatment options are presently available; (4) the impairment of cell's protein turnover machinery leads to the deposition of extracellular and intracellular protein aggregates, which in turn induce NI with the recruitment of cells of the immune system (microglia) contributing to neurotoxicity and degeneration of neurons (Figure 1); (v) diagnosis is based on clinical symptoms appearing at a late stage in the course of the disease: in PD, for example, clinical signs appear when over 50% of disease-specific neurons (nigrostriatal projections) are already damaged.
As reviewed by Rivest (2009), Heneka et al (2010b), Perry et al (2010), and Glass et al (2010), in AD, Aβ forms extracellular aggregates that activate microglia through TLRs and receptor for advanced glycation end products (Figure 1), leading to the activation of nuclear factor-κB and activator protein with subsequent production of reactive oxygen species and release of cytokines (TNF-α, IL-1β, and IL-6) that stimulate astrocytes, which amplify the inflammatory/neurotoxic effects. All these events eventually contribute to neuronal cell death, which in turn leads to the release of ATP with further activation of microglial cells through purinergic P2X7 receptors. However, α- and β-secretases have nuclear factor-κB binding sites in their promoters, and the expression of proinflammatory cytokines appears upregulated in neurons, serving for more production of Aβ in a ‘vicious cycle’ between inducers, transducers, and effectors. In PD, aggregates of α-synuclein (α-SYN) form intermediate-state oligomers that, when released from neurons, activate microglia through TLR-independent mechanisms. In ALS, aggregates of SOD1 (superoxide dismutase 1) can induce inflammatory responses by microglia through TLR2 and CD14 (Glass et al, 2010). However, some authors have stated that extracellular amyloid deposits or chronic neurodegeneration on their own are not able to elicit a robust proinflammatory response (Perry et al, 2010).
Although microglia activation seems to be induced in a disease-specific manner, there is a remarkable convergence in the mechanisms of sensing, transduction, and amplification of the inflammatory process. It should be pointed out that, while sustained inflammatory responses involving microglia and astrocytes are likely to contribute to disease progression, microglia also have a neuroprotective role by mediating the clearance of Aβ (Figure 1). Therefore, a still unresolved question is how to control and modulate microglial activity, in a safe and efficient manner, to slow down or reverse the course of ND. A better understanding of the brain's innate immune response will help to develop strategies, e.g., based on synthetic TLR agonists, to selectively activate neuroprotective microglia functions while avoiding detrimental effects on neurons (Rivest, 2009).
Positron-emission tomography and MRI have contributed a broad spectrum of imaging findings in ND: (1) altered cerebral glucose consumption (AD); (2) altered neuronal transmission within the cholinergic, dopaminergic, serotonergic, and other neurotransmitter systems (AD and PD); (3) accumulation of Aβ and tau proteins (AD and PD); (4) degeneration of central motor neurons (ALS) (Berti et al, 2011).
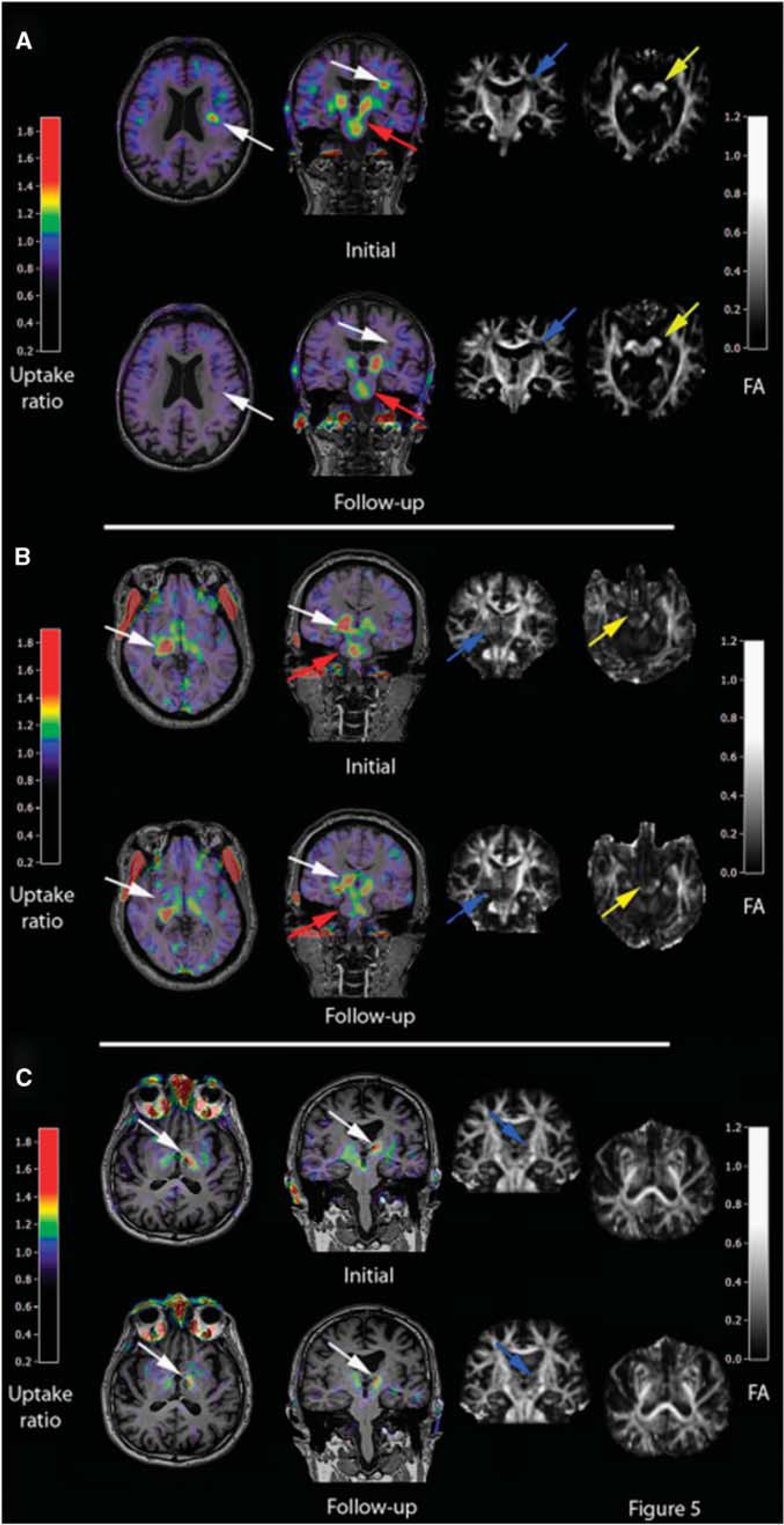
Imaging studies of NI in ND have mostly focused on microglial activation. As pointed out in Figure 1, Aβ deposition elicits a vicious cycle of NI and neuronal destruction, where microglia have the central role. Positron-emission tomography with [11C](R)-PK11195 showed quantitatively the in-vivo microglial activation involving the entorhinal, temporoparietal, and cingulate cortices (n=8 AD, n=1 MCI, n=6 normal) (Cagnin et al, 2001a). In patients with frontotemporal dementia, PET showed [11C](R)-PK11195 binding in the frontal, medial temporal, and subcortical regions (Cagnin et al, 2004). In APP/PS1 mice, [3H](R)-PK11195 binding correlated with the microglial activation determined by immunohistochemistry (Venneti et al, 2009). Kinetic modeling has shown specific binding potentials (BPs) of [11C](R)-PK11195 in the human brain (Schuitemaker et al, 2007) using a simplified reference-tissue model (Kropholler et al, 2007). The inclusion of an additional vascular component effectively modelized disease-specific vascular changes but amplified the BP more in AD than in controls because of a decrease in tracer binding to the vasculature in the AD cohort (Tomasi et al, 2008). It should be pointed out that the cerebellum as a reference tissue creates problems because of its proximity to vascular sinuses containing TSPO. Therefore, several teams are exploring clustering methods using a database of tissue kinetics to identify healthy gray-matter voxels that are used as a reference (Turkheimer et al, 2007). In double tracer studies of patients with AD using [11C]PIB together with [11C](R)-PK11195, the distribution of the amyloid load and the microglial activation were correlated with the cognitive status (n=13) (Edison et al, 2008). [11C]PIB-PET revealed a significant twofold increase in the amyloid load in frontal, temporal, parietal, occipital, and cingulate cortices, while [11C](R)-PK11195-PET detected significant 20% to 35% increases in microglial activation in the same cortical areas. Importantly, the mini mental state examination scores in AD patients correlated with the levels of cortical microglial activation but not with the amyloid load, supporting the view that microglia activation is related to disease activity and progression (Edison et al, 2008) even at a stage of presymptomatic disease such as MCI (Okello et al, 2009; Figure 6). A recent study explored the relationship between Aβ accumulation and NI in relation to glucose metabolism in patients with early AD (n=11) (Yokokura et al, 2011). A negative correlation was found between dementia score and the [11C](R)-PK11195 BP, but not between score and [11C]PIB uptake in the limbic, precuneus and prefrontal regions. Direct comparisons showed a significant negative correlation between [11C](R)-PK11195 and [11C]PIB BPs in the posterior cingulate cortex, the region that manifested the most severe reduction in [18F]FDG uptake. The lack of coupling between microglial activation and amyloid deposits may indicate that Aβ accumulation may not always be the primary cause of microglial activation, but rather the negative correlation present in the posterior cingulate cortex suggests that microglia can show higher activation during the production of Aβ in early AD (Yokokura et al, 2011). Considering that norepinephrine, which is decreased in depression, has been shown to suppress Aβ-induced cytokine and chemokine production and to increase microglia migration and phagocytosis of Aβ (Heneka et al, 2010a), while antidepressants have been shown to limit amyloid brain deposition in mice through decrease in TNF-α (Chavant et al, 2010), it cannot be excluded that NI could be the missing link between aging depression and an increased risk of ND (Frank-Cannon et al, 2009).
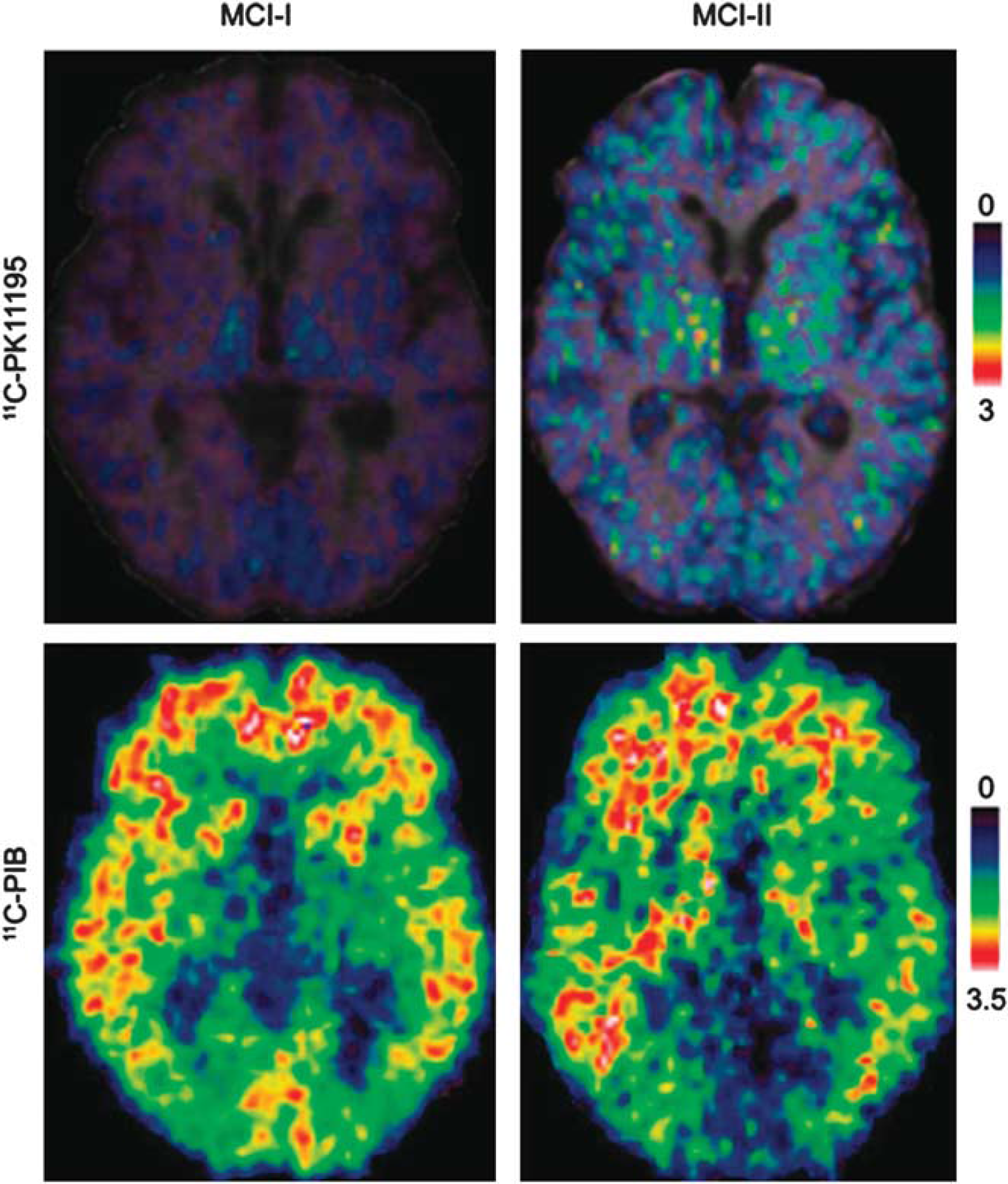
Early detection of amyloid-β as damage-associated molecular pattern (DAMP) and microglia activation in patients with mild cognitive impairment by positron-emission tomography (PET). [11C]-(R)-PK11195 binding potentials (BPs) and corresponding [11C]PIB ratio images in two patients with PIB-positive mild cognitive impairment (MCI). MCI-I has normal PK binding and MCI-II has increased PK binding suggesting that Aβ deposition may be related but also unrelated to microglia activation at very early disease stages. The detection of microglial activation in patients with MCI suggests that anti-inflammatory therapies may be relevant to the prevention of AD. (Figure reprinted with permission from Okello et al, 2009.)
Studies in experimental animals and patients with PD showed that NI, as determined by [11C](R)-PK11195-PET, is also a significant component of progressive dopaminergic degeneration (Cicchetti et al, 2002; Gerhard et al, 2006), even though it may be difficult to quantify in the clinical situation (Bartels et al, 2010). Nevertheless, ‘imaging-guided’ drug intervention studies have been initiated, e.g., in patients with multiple system atrophy focusing on minocycline-induced alterations of microglia activation in vivo (Dodel et al, 2010).
Microglial activation in ALS has been studied only in a very limited number of patients (n=10) (Turner et al, 2004). Increased binding was found in motor cortex, pons, dorsolateral prefrontal cortex, and thalamus, and a significant correlation between binding in the motor cortex and the burden of clinical upper motor neuron signs was shown (r=0.73, P=0.009). These findings indicate that cerebral microglial activation during the evolution of ALS can be detected in vivo, and support the view that cerebral pathology is widespread in this disease (Turner et al, 2004). In an elegant imaging-guided study by Keller et al, it was shown in the glial fibrillary acidic protein-luc/SOD1G93A reporter mouse model of ALS that interfering with microglia activity by means of minocycline at various disease stages either ameliorates (at early stages) or enhances (at later stages) disease activity and symptoms. Pretreatment with minocycline delayed disease progression and increased mean survival, whereas minocycline administered at later stages of disease was associated with significant alterations in astrocyte and microglia activation profiles (Keller et al, 2011). Using another mouse model of ALS (SOD1G93A) with labeled projection neurons and labeled microglia/macrophages, in-vivo imaging by two-photon laser scanning microscopy compared the role of microglia/macrophage-related NI in the CNS and peripheral nervous system and showed the disease-driving role of highly reactive microglia cells in preclinical disease stages (Dibaj et al, 2011).
Epilepsy: As has been nicely reviewed recently (Vezzani et al, 2011), glial cells are implicated in a number of molecular mechanisms of seizure precipitation and recurrence: (1) alterations in the phenotype and function of activated microglia and astrocytes; (2) modifications in potassium and water channels; (3) alterations of glutamine/glutamate cycle and glutamate receptor expression; (4) release of neuromodulatory molecules (gliotransmitters and neurotrophic factors); and (5) induction of neuroinflammatory molecules (cytokines, chemokines, prostaglandins, complement factors, and cell adhesion molecules). This review pointed out that brain injury and proconvulsant events can activate microglia and astrocytes to release a number of proinflammatory mediators initiating a cascade of neuroinflammatory processes in the brain (Vezzani et al, 2011). So far, imaging of NI in the context of epilepsy has only been performed in single patients. [11C]-(R)-PK11195 uptake was observed in regions of epileptic foci and focal cortical dysplasia as determined by high-resolution MRI, electroencephalography recording and intraictal and interictal [18F]FDG-PET (Butler et al, 2011). These findings confirm results from invasive studies that revealed a specific and persistent increase in the numerical density of activated microglia within dysplastic regions of patients with epilepsy, and support the view that the inflammatory response and proinflammatory molecules contribute to epileptogenicity of focal cortical dysplasia (Boer et al, 2006).
Encephalitis: T2 and fluid attenuation inversion-recovery MRI sequences are the imaging methods of choice for detection with a high sensitivity of single changes in the temporal lobe caused by HSV-1 infection/reactivation. An enzymatic assay of thymidine kinase of HSV-1 (HSV-1-tk) using radiolabeled antiviral drugs was proposed 30 years ago for specific detection of active virus replication in vivo (Saito et al, 1982). This enzymatic assay was later developed for imaging transduced gene expression in vivo and initiated together with other developments the exciting field of ‘molecular genetic imaging’ (Jacobs et al, 1999; Tjuvajev et al, 1995), which has been applied to clinical situations (Jacobs et al, 2001b). This imaging technology can also assess in-vivo replication of recombinant HSV-1 vectors in vivo (Jacobs et al, 2001a).
Apart from approaches to directly image herpes virus particles in vivo, microglial markers have been used to quantify herpes encephalitis-induced microglia activation in vivo (Cagnin et al, 2001b; Doorduin et al, 2010). Similarly to studies in stroke (Figure 3), it was shown in patients with HSV-1 encephalitis (n=2) that [11C](R)-PK11195-PET uptake increases significantly within the affected limbic system, and additionally in areas connected to the limbic system by neural pathways such as the lingual gyrus in the occipital lobe and the inferior parietal lobe, where uptake persists for many months (>12) after antiviral treatment (Cagnin et al, 2001b). Interestingly, cortical areas showing an early high [11C](R)-PK11195 binding subsequently underwent atrophy, underlining the neurotoxic effects of microglia (Figure 1). It should be pointed out that, in contrast to HSV-1 encephalitis, [11C](R)-PK11195-PET studies in patients with human immunodeficiency virus (HIV) encephalitis were unable to detect significant activation of microglia in vivo, indicating that either [11C](R)-PK11195 PET assessment is insensitive to the degree of macrophage activation in the HIV-associated minor neurocognitive impairment, or that macrophage activation is not the pathological substrate of this neurologic condition (Wiley et al, 2006). The development of improved TSPO tracers may give an answer to this question (Doorduin et al, 2009b; Venneti et al, 2008a,2008b). Finally, in an experimental macaque model system, activated macrophages in lentiviral encephalitis showed an increase in [3H](R)-PK11195 binding in a phosphoinositide 3 kinase-dependent manner (Venneti et al, 2007).
Gliomas: Primary neoplasms are the second most common cause of death from an intracranial disease. Imaging based on PET and MRI has a crucial role for diagnosis, therapy guidance and in the recurrent situation (Dhermain et al, 2010). Whereas PET markers for increased amino-acid and nucleoside metabolism are nowadays used on a regular basis by many centers, TSPO could also be an exciting target to study the interrelation between glioma and microglial cells in vivo (Buck et al, 2011; Takaya et al, 2007). In patient samples, TSPO suppression in glioma-infiltrating microglia suggested that TSPO might have a relevant role in modulating the antitumor inflammatory response in astrocytic tumors (Takaya et al, 2007). The expression of TSPO has been shown in experimental glioma models and a direct visualization of the location and extent of the tumor was observed by microPET imaging with TSPO radiotracers by several groups (Buck et al, 2011; Tang et al, 2012; Winkeler et al, 2012).
Conclusion
Neuroinflammation is a complex orchestrated response to various stimuli aiming toward tissue preservation and restoration, which, under pathological conditions, paradoxically increases tissue damage and disease activity. In most neurologic diseases, the neuroinflammatory response determines tissue outcome. Therefore, a precise and reliable detection of NI is of great fundamental and clinical interest. Molecular imaging aims at deciphering the complex interplay of various cellular and molecular actors of NI in vivo. Presently, its main applications are the direct visualization of recruitment of specific cells from the periphery into the brain by two-photon microscopy (e.g., T cells) and MRI (macrophages) or the activation of microglia by PET and SPECT imaging. However, the long-term goal of this research aims to decipher imaging markers for NI which will guide the therapeutic interventions to alter the neurotoxic inflammatory cascade, prevent tissue damage, and enhance tissue repair. One key to success is the understanding and noninvasive revealing of the various microglial phenotypes during the course of diseases, to distinguish ‘neurotoxic’ from ‘neuroprotective’ microglia and to document the action of treatments inhibiting the former and stimulating the latter for an improved tissue outcome.
Footnotes
Acknowledgements
The review was initiated by an extensive process of scientific interactions with many colleges within the research carried out during the EU FW6 network of excellence program DIMI (http://www.dimi.eu) and within the design of the EU FW7 large scale integrating project INMiND (http://www.uni-muenster.de/InMind). The authors thank all colleagues within these research programs for sharing their knowledge and ideas. The authors also thank Prof. Ulrich Dirnagl (Berlin), Dr Hervé Boutin (Manchester), Prof. Adriaan Lammertsma (Amsterdam), and Prof. Gitte Knudsen (Copenhagen) to present parts of their research topics within the field of neuroinflammation during a dedicated Session organized by the European Society for Molecular Imaging (ESMI) at the Brain2011 conference in Barcelona. Moreover, the authors thank Nina Gerigk (EIMI, Münster) for the design of ![]() and Dr Yannic Waerzeggers (EIMI, Münster) for proof reading of the manuscript.
and Dr Yannic Waerzeggers (EIMI, Münster) for proof reading of the manuscript.
Disclosure/conflict of interest
The authors declare no conflict of interest.