
Abstract
The clearance of amyloid beta (Aβ) from the brain represents a novel therapeutic target for Alzheimer's disease. Conflicting data exist regarding the contribution of adenosine triphosphatebinding cassette transporters to the clearance of Aβ through the blood-brain barrier. Therefore, we investigated whether Aβ could be a substrate for P-glycoprotein (P-gp) and/or for breast cancer resistance protein (BCRP) using a human brain endothelial cell line, hCMEC/D3. Inhibition of P-gp and BCRP increased apical-to-basolateral, but not basolateral-to-apical, permeability of hCMEC/D3 cells to 125l Aβ 1–40. Our in vitro data suggest that P-gp and BCRP might act to prevent the blood-borne Aβ 1–40 from entering the brain.
Introduction
Increased levels of neurotoxic amyloid beta (Aβ) peptides of 1–40 and 1–42 amino acids in length (Aβ 1–40, 1–42) are thought to be central in the pathogenesis of Alzheimer's disease (AD) (Hardy, 2006). A potential therapeutic treatment for AD is to increase the clearance of Aβ from the brain thus, reducing the amyloid burden. Brain endothelial cells (BECs) at the blood-brain barrier (BBB) express multiple transporters for Aβ, which lead to Aβ influx into (RAGE (receptor for advanced glycation end products), megalin) and efflux from (LRP-1 (low-density lipoprotein receptor related protein)) the central nervous system (Zlokovic, 2008). BECs also express the adenosine triphosphate-binding cassette efflux transporters, P-glycoprotein (P-gp), and the breast cancer resistance protein (BCRP), which have a similar substrate overlap, and act to prevent unwanted blood-borne signalling molecules from entering the brain (Loscher and Potschka, 2005). P-gp has been recently described as a potential Aβ transporter (Cirrito et al, 2005; Kuhnke et al, 2007; Lam et al, 2001), although evidence is conflicting (Ito et al, 2006; Nazer et al, 2008). To date, there have been no reported investigations regarding the interactions between Aβ and P-gp or BCRP in human BECs in vitro. Therefore, we examined the role of each efflux transporter on Aβ 1–40 transport using a physiologic concentration of 125I Aβ 1–40 in an immortalised human BEC line, hCMEC/D3 cells.
Materials and methods
Cell Culture
hCMEC/D3 cells were cultured in EBM-2 medium (Lonza, Slough, Wokingham, UK), which was supplemented with vascular endothelial growth factor (VEGF), insulin-like growth factor (IGF), basic fibroblast growth factor (bFGF), hydrocortisone, ascorbate, gentamycin, and 2.5% fetal bovine serum (FBS). hCMEC/D3 cells were cultured on collagen-coated (Sigma-Aldrich, Gillingham, Dorset, UK; 0.005% w/v, 1 h at room temperature) 24-well tissue culture plates in a humidified atmosphere at 37°C in 5% CO2. For permeability experiments, transwell polyester membrane inserts (Corning Costar, High Wycombe, Buckinghamshire, UK; 0.4-μm pore, 12-mm diameter) were first coated with collagen and then with fibronectin (Sigma-Aldrich; 5 μg/mL, 1 h). hCMEC/D3 cells were grown to confluence (∼1 × 105cells/cm2) with a culture media change every 2 to 3 days and incubated for 2 days after confluence before treatment.
125I Aβ 1–40 Net Uptake by hCMEC/D3 Cells
hCMEC/D3 cells were incubated for 0.5, 1, 3, or 24 h with 0.1 nmol/L 125I Aβ 1–40 (Bachem, Weil am Rheim, Germany; 1,110 Ci/mmol). For some experiments, hCMEC/ D3 cells were incubated for 30 mins with 0.1 nmol/L of 125I Aβ 1–40 and 2 μmol/L vinblastine (a competitive P-gp and multi-drug resistance protein-1 (MRP-1) inhibitor; Sigma-Aldrich), 300nmol/L of tariquidar (a specific P-gp inhibitor; a kind gift from Xenova, Slough, Berkshire, UK), 2 μmol/L of fumitrimorgan C (FTC, a competitive BCRP inhibitor; Axxora, Nottingham, Nottinghamshire, UK), or a dimethyl sulfoxide (DMSO) vehicle control. For all samples, the 125I Aβ remaining in the media and in trypsinised hCMEC/D3 cells was measured using a γ-counter (Wallac Wizard, Perkin Elmer, Cambridgeshire, UK). For these and for the subsequent experiments using 125I Aβ 1–40, culture media or whole cell lysates were precipitated with 15% trichloroacetic acid overnight at 4°C, and radioactivity corresponding to free 125I (∼1 to 2%) was subtracted from total counts resulting in a measure of peptide-bound 125I. The total amount of protein in each sample was determined using a DC protein assay (Bio-Rad, Hemel Hempstead, Hertfordshire, UK) and the net uptake of 125I Aβ by hCMEC/D3 cells was related to the total amount of protein in cell suspension (fmol/mg).
125I Aβ 1–40 Efflux from hCMEC/D3 Cells
hCMEC/D3 cells were preloaded with 0.1 nmol/L of 125I Aβ 1–40 for 3 h, the culture media removed, and the levels of 125I Aβ 1–40 counted. hCMEC/D3 cells were incubated in a fresh culture medium containing 300 nmol/L of tariquidar, 2 μmol/L vinblastine, 2 μmol/L FTC, or vehicle alone for 7.5, 15, 30, 60, or 180 mins at 37°C. A further treatment was also collected, whereby preloaded cells were incubated at 4°C for 180 mins. For some experiments, 125I Aβ 1–40 preloaded hCMEC/D3 cells were incubated in fresh culture media containing vinblastine (2 μmol/L), tariquidar (300 nmol/L), FTC (2 μmol/L), or DMSO for 10 mins at 37°C. The amount of protein per milligram of total protein was calculated as described above.
125I Aβ 1–40 Permeability of hCMEC/D3 Cells
Confluent hCMEC/D3 cells grown on inserts were incubated for 30 mins with 0.1 nmol/L of 125I Aβ and 0.1 mmol/L 14C Inulin (1 to 3 μCi/mg; MP Biomedicals, Solon, Ohio, USA) in either the apical or basolateral chamber in the presence of 2 μmol/L of vinblastine, 300 nmol/L tariquidar, 2 μmol/L FTC, or DMSO vehicle control. The Aβ clearance quotient in the apical-to-basolateral (CQAP) and basolateral-to-apical (CQBL) directions, described by Nazer et al, 2008, was calculated with modifications using the 125I and 14C counts per minute (c.p.m.) measured by a γ- and β-counter (Wallac Trilux, Perkin Elmer), using the following formulas:
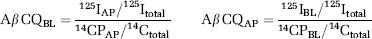
where 125Itotal is the total c.p.m. for the apical and basal compartments, as well as for those remaining in trypsinised cells and inserts. 14Ctotal is the total inulin disintegrations per minute (d.p.m.) for the apical and basolateral compartments. 125IAP and 14CAP are the c.p.m. and d.p.m., respectively, in the apical compartment whereas 125IBL and 14CBL represent c.p.m. and d.p.m., respectively, in the basolateral compartment.
Statistical Analysis
All data are represented as means ± s.e.m., and the number of experiments, n, indicated each time. For all experiments, a paired Student's t-test was used.
Results
125I Aβ 1–40 Net Uptake by hCMEC/D3 Cells
Initially, the net uptake of 0.1 nmol/L 125I Aβ 1–40 was investigated as an indication of all cellular processes regulating Aβ 1–40 levels in hCMEC/D3 cells. The Aβ 1–40 net uptake refers to all processes that influence Aβ 1–40 cellular levels in hCMEC/D3 cells, including those regulating influx (receptor-mediated internalisation, diffusion, fluid-phase endocytosis, and any ‘sticking’ to the cell membrane) and efflux (efflux transport, vesicular release, diffusion, and dissociation from the plasma membrane). 125I Aβ 1–40 net uptake by hCMEC/D3 cells was time-dependent reaching saturation between 3 and 6 h, with a t1/2 (time taken to achieve half maximal uptake) of ∼ 35 mins (Figure 1A). Tariquidar (300 nmol/L), vinblastine (2 μmol/L), and FTC (2 μmol/L) increased the net uptake of 0.1 nmol/L 125I Aβ 1–40 after a 30-min incubation by 15%, 8%, and 17% of control levels, respectively. However, only that of tariquidar and vinblastine reached statistical significance (Figure 1B). These results imply that 125I Aβ 1–40 cellular levels may be influenced by the efflux activity of P-gp, and possibly also that of BCRP.
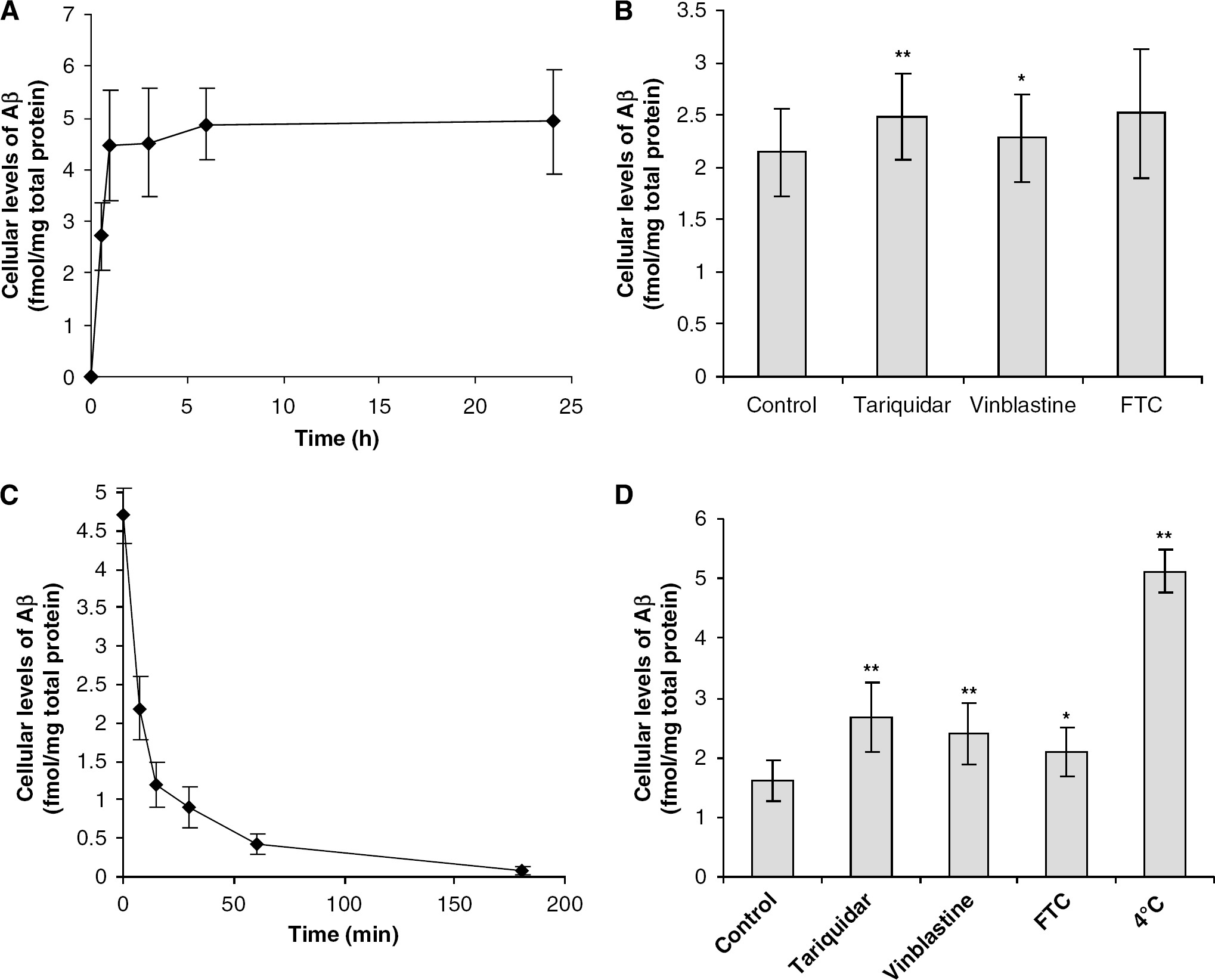
Net uptake and efflux of Aβ 1–40 by hCEMC/D3 cells. The net uptake of 0.1 nmol/L 125l Aβ 1–40 by hCMEC/D3 cells (
125I Aβ 1–40 Efflux from hCMEC/D3 Cells
To further explore the possibility that 125I Aβ 1–40 transport out of hCMEC/D3 cells might be mediated by P-gp or BCRP, we investigated Aβ 1–40 efflux from preloaded hCMEC/D3 cells (3 h, 0.1 nmol/L) (Figure 1C and ID). 125I Aβ 1–40 efflux from hCMEC/ D3 cells was time-dependent when compared with maximal cellular Aβ 1–40 levels (preloaded cells incubated at 4°C), with a t1/2 of 8.1 ± 1.3 mins, whereas incubation with tariquidar, vinblastine, and FTC increased the t1/2 by 19.6 ± 5.5%, 19.0 ± 1.0%, and 20.0 ± 0.5%, respectively. After a 10-min incubation at 37°C, tariquidar, vinblastine, and FTC increased the cellular levels of 125I Aβ 1–40 by 66%, 49%, and 31%, respectively, compared with control levels. The results indicate that P-gp and BCRP contribute to the efflux of Aβ 1–40 from hCMEC/D3 cells.
hCMEC/D3 Cell Permeability to 125I Aβ 1–40
The permeability of hCMEC/D3 cells to Aβ 1–40 is a consequence of paracellular diffusion and intracellular processes that include both influx and efflux mechanisms. Therefore, the permeability of hCMEC/ D3 cells grown on inserts to 0.1 nmol/L of 125I Aβ 1–40 in the presence of tariquidar (300 nmol/L), vinblastine (2 μmol/L) and FTC (2 μmol/L), or DMSO control was investigated over a period of 30 mins (Figure 2). The Aβ CQAP of hCMEC/D3 cells to 125I Aβ 1–40 was increased by 63% with tariquidar (300 nmol/L), 49% with vinblastine (2 μmol/L), and 46% by FTC (2 μmol/L) when compared with control levels. In contrast, the Aβ CQBL was unaltered by tariquidar, vinblastine, or FTC. These results imply that P-gp and BCRP act to prevent the apical-to-basolateral transport of physiologic concentrations of 125I Aβ 1–40. By contrast, P-gp and BCRP may not mediate the Aβ basolateral-to-apical transport.
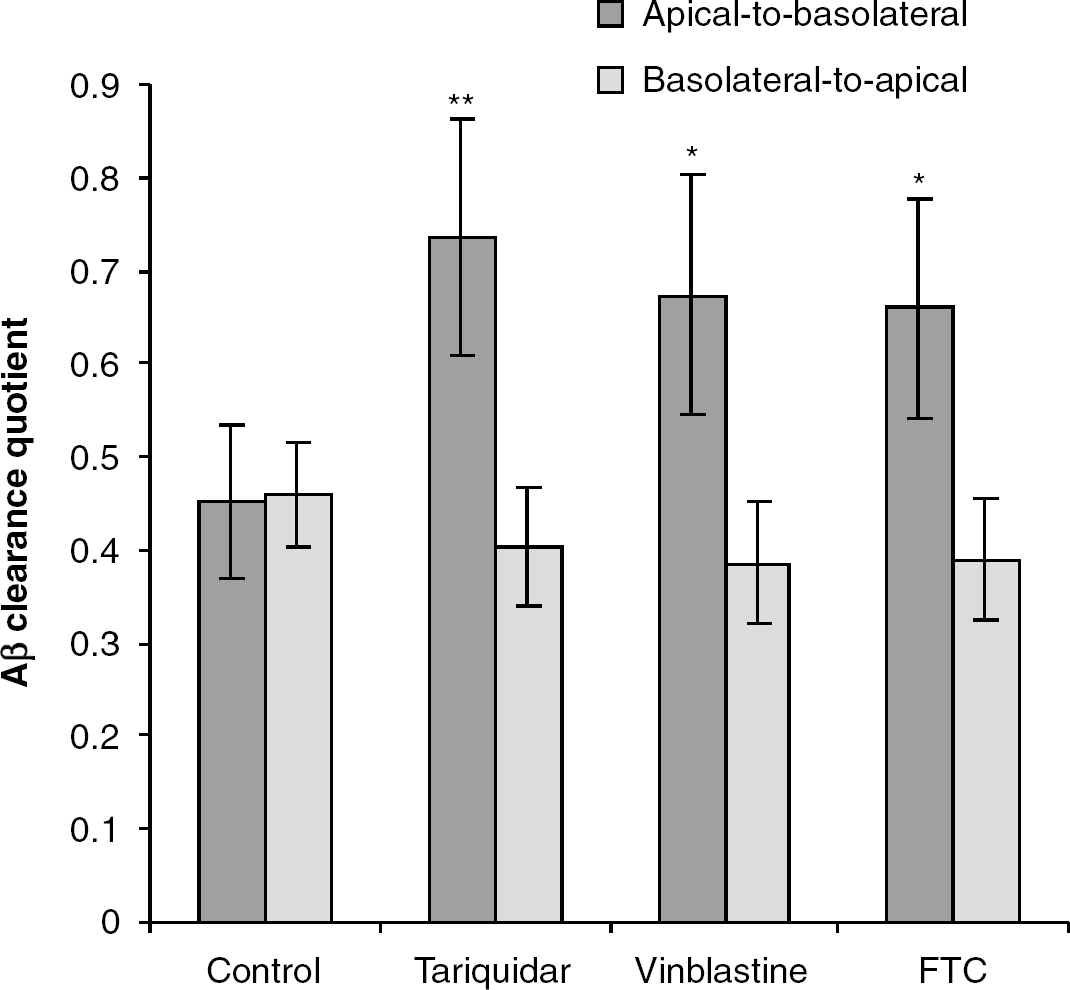
Permeability of hCMEC/D3 cells to Aβ 1–40. Cells were grown to confluence on transwell filter inserts and incubated with 0.1 nmol/L 125l Aβ 1–40, 0.1 mmol/L 14C inulin and tariquidar (300 nmol/L), vinblastine (2 μmol/L), FTC (2 μmol/L) or a DMSO vehicle control for 30 mins. The 125l and 14C c.p.m. were measured and the Aβ 1–40 clearance quotient in the apical-to-basolateral and basolateral-to-apical directions calculated. Data represent mean ± s.e.m.; n = 8. *P < 0.05, **P < 0.01, comparing samples treated with inhibitors and untreated controls, using a paired Student's t-test.
Discussion
The results of this study show that, at physiologic concentrations, P-gp and BCRP inhibition results in an increased net uptake and in a decreased efflux of 125I Aβ 1–40 from hCMEC/D3 cells. Brain endothelial cells express proteins capable of Aβ transport into the brain, most notably RAGE and megalin, and out of the brain, namely LRP-1 (Zlokovic, 2008). Some studies have indicated that P-gp may also influence Aβ transcytosis across BECs but, to date, there are no studies investigating the role of P-gp or BCRP in Aβ transport by human BECs in vitro. Our results are consistent with earlier in vitro binding data showing possible interactions between P-gp and Aβ using nonhuman mammalian cells. Initially, Aβ 1–40 and 1–42 were described to compete for the uptake of 3H-colchicine in Chinese hamster ovary cells overexpressing multi-drug resistance-1 (MDR1) (Lam et al, 2001) and with rhodamine 123 in porcine kidney epithelial cells transfected with human MDR1 (LLC-MDR1 cells) membrane vesicles (Kuhnke et al, 2007). Furthermore, P-gp inhibitors, cyclosporin A and verapamil, were found to reduce the uptake of fluoresceinisothiocyanate (FITC) conjugated Aβ 1–40 and Aβ 1–42 in LLC-MDR1 membrane vesicles. We sought to use a more physiologically relevant model of Aβ 1–40 transport using human BECs grown on inserts, the results of which indicate that BCRP and P-gp both play a role in preventing the apical-to-basolateral, but not basolateral-to-apical, permeability of hCMEC/D3 cells to 125I Aβ 1–40. Support for the theory that P-gp efflux activity restricts the BBB permeability to Aβ has been obtained from cell lines, which are not endothelial in origin and has produced conflicting results. LLC-MDR1 cells have enhanced basolateral-to-apical, but not apical-to-basolateral, permeability to FITC-Aβ 1–40/42 (5 μmol/L, 1 h) compared with wild-type LLC cells, which was inhibited by 10 μmol/L of cyclosporin A (Kuhnke et al, 2007). In contrast, the basolateral-to-apical permeability of MDCK-MDR1 (Madin-Darby canine kidney epithelial cells overexpressing human MDR1) is not increased compared with wild-type MDCK cells after incubation with 1 nmol/L of 125I Aβ 1–40 for 24 and 48 h (Nazer et al, 2008). These apparently contradicting results may reflect differences in the plasma membrane distribution of efflux transporters between different cultured epithelial cell lines. Although a direct interaction between Aβ 1–40 and P-gp or BCRP by, e.g., coimmunoprecipitation techniques remains to be shown, our results using a human BEC line are consistent with a polarised expression of P-gp and BCRP on the apical surface of human cerebral microvessels preventing Aβ 1–40 transcytosis (Cooray et al, 2002; Seetharaman et al, 1998). Interestingly, 125I Aβ 1–40 has been observed to bind to primary human BECs only on the apical membrane and appears to be selectively transported towards the basolateral membrane (Mackic et al, 1998). However, 65% of the BEC apical binding was found to be attributed to RAGE. It should be noted that astrocytes may influence the polarity of Aβ transporters, and that the results from that study were obtained in the absence of astrocytes or astrocyte-conditioned media. In addition, RAGE-Aβ interactions are important for the migration of monocytes/macrophages across the BBB (Giri et al, 2000). Although RAGE might mediate the apical-to-basolateral transport of Aβ, our data suggest that P-gp and BCRP on the apical surface of endothelial cells may selectively restrict the apical-to-basolateral permeability of the BBB to Aβ 1–40. By contrast, the basolateral-to-apical transport of Aβ across BECs, previously shown to be mediated by LRP-1, according to our results, is not affected by P-gp or BCRP inhibition. In vivo, Tg2576 transgenic mice, which harbour the human APP Swedish mutation gene, when treated with a P-gp inhibitor (XR9576) have increased brain interstitial fluid (ISF) levels of Aβ (Cirrito et al, 2005). These results may be on account of a consequence of an increased blood-to-brain Aβ transport rather than because of a decreased Aβ clearance from the brain. Indeed, the brain efflux of human Aβ microinjected intracerebrally was reported to be unaltered in mice treated with P-gp inhibitors, verapamil and quinidine (Ito et al, 2006). In vivo, the specific contribution of BBB Aβ transporters is difficult to interpret as brain resident cells, such as astrocytes (Pardridge et al, 1997), and cells in the periphery, such as epithelial cells (Schinkel and Jonker, 2003), are also able to express P-gp. Furthermore, the contribution of P-gp in protecting the brain from plasma Aβ species is often overlooked in vivo.
Our in vitro results show that P-gp and BCRP restrict the apical-to-basolateral permeability of hCMEC/D3 cells to Aβ 1–40, suggesting a role for these efflux transporters in preventing blood-borne Aβ 1–40 from entering the brain, but not in transporting the central nervous system Aβ 1–40 into blood. However, in vivo studies need to be carried out to confirm these observations, and also to verify whether other Aβ species are transported similarly. Interestingly, the increased plasma levels of Aβ are reported in AD patients compared with that in controls by as much as 300% (Lichtlen and Mohajeri, 2008; Sagare et al, 2007). Therefore, P-gp, and to a lesser extent BCRP, might act to prevent the plasma Aβ pool from entering the brain. Many drugs that are in therapeutic use today, such as lipid lowering agents and antidepressants, are transported by P-gp and in principle might act as competitive substrates with Aβ for P-gp at the BBB. Therefore, caution should be applied when prescribing drugs, which are P-gp substrates, to AD patients with increased plasma levels of Aβ.
Footnotes
The authors declare no conflicts of interest.