
Abstract
Mild hypothermia renders potent neuroprotection against acute brain injury. Recent reports show that adenosine 5'-monophosphate (AMP) plays a role in thermoregulation and induces hypothermia in mice. Therefore, this study sought to determine whether AMP induces hypothermia in rats and to study its collective effects on cerebral ischemia induced by 2-h middle cerebral artery occlusion. An intraperitoneal injection of AMP induced hypothermia dose-dependently. At the dose of 4 mmol/kg, AMP induced promising mild hypothermia for 2.5 h. Unexpectedly, the AMP-induced hypothermia failed to reduce infarct volume after brain ischemia; instead, it exaggerated the ischemic damage, indicated by an increased infarct volume, as well as increased incidences of hemorrhagic transformation, seizure, and animal death. Physiologic parameter monitoring revealed that AMP causes profound hypotension, leading to cerebral hypoperfusion. Furthermore, AMP administration resulted in severe hyperglycemia, metabolic acidosis, and hypocalcemia. In addition, western blots showed early dephosphorylation and degradation of AMP-activated kinase in the ischemic cortex in AMP-treated rats. Taken together, our findings suggest that AMP induces hypothermia in rats, probably by limiting cellular access to glucose. However, the potential neuroprotection of AMP-mediated hypothermia against ischemia was overwhelmed by the detrimental effects of hypotension and hyperglycemia, thus making AMP an unlikely agent for inducing hypothermia to protect the brain against ischemic injury.
Introduction
Acute brain injury occurs after cerebral ischemia, subarachnoid hemorrhage, or traumatic brain injury, leading to permanent neuronal loss. In the fight against acute neuronal damage, mild hypothermia emerges as a potent performer with neuroprotective effects. Mild hypothermia, with temperatures of 32°C to 34°C, protects the brain against injuries induced by ischemia (Maier et al, 1998; Miyazawa et al, 2003; The Hypothermia after Cardiac Arrest Study Group, 2002; Torup et al, 2003), subarachnoid hemorrhage (Thome et al, 2005), and traumatic brain injury (Peterson et al, 2008).
Among these acute brain injuries, ischemic stroke brings about the greatest social and economic burden, because it is the leading cause of long-term disability and the third leading cause of human death. Although hypothermia is very effective in attenuating ischemic brain injury in lab animals (Luo et al, 2007; Taniguchi et al, 2005) and human patients with global cerebral ischemia (The Hypothermia after Cardiac Arrest Study Group, 2002), the methods used to induce hypothermia are usually external and inconvenient. These methods include ice (Luo et al, 2007; Maier et al, 1998), alcohol spray (Zhao et al, 2005), and special devices (Clark and Colbourne, 2007; Taniguchi et al, 2005; The Hypothermia after Cardiac Arrest Study Group, 2002), none of which are practical in the emergency setting because their application requires that the subjects be anesthetized. Therefore, it will be promising if mild hypothermia can be induced in nonanesthetized lab animals and humans using biomolecules with thermo-regulating effects.
Adenosine 5'-monophosphate (AMP), a natural molecule of adenosine triphosphate (ATP) metabolism, was recently shown to have a thermoregulation function (Zhang et al, 2006). Administration of exogenous AMP rapidly induced mild-to-moderate hypothermia in nonhibernating mice (Swoap et al, 2007; Zhang et al, 2006). If AMP can be shown to cause hypothermia in other species of animals and in humans, and if it shows a neuroprotective effect, it would be possible to use mild hypothermia as a routine practice in both research and clinical settings against acute brain injury.
The purpose of this study was to determine whether AMP induces hypothermia in rats and to study its collective effects on focal cerebral ischemia. Our results show that AMP induces hypothermia in rats in a dose-dependent manner, with 4 mmol/kg body weight, which is a favorable dose for the desired duration of mild hypothermia. However, AMP unexpectedly confers a detrimental rather than a beneficial effect on the ischemic outcomes in rats. It turns out that AMP also causes hyperglycemia and hypotension, which may overwhelm the beneficial effects of AMP-induced hypothermia. Thus, our results exclude AMP as a cryogen for the induction of hypothermia to attenuate ischemic injury in the brain.
Materials and methods
Animal and Physiologic Parameter Monitoring
All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and carried out in accordance with the NIH's Guide for the Care and Use of Laboratory Animals. The animals used were male adult Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA, USA) weighing 280 to 310 g. The rats were housed at 21°C to 24°C with a 12:12 h light-dark cycle, with free access to food and water unless indicated otherwise.
Core body temperature (CBT) was measured rectally using an Omega 450 AET Thermocouple Thermometer (OMEGA Engineering, Inc., Stamford, CT, USA). The measurement was taken every 30 mins while the rats were awake unless indicated otherwise. The brain temperature was monitored using an Omega HH-25TC Thermometer with a 29-Ga thermocouple (OMEGA Engineering), as described earlier (Karibe et al, 1994; Luo et al, 2007). In brief, the rats were anesthetized with isoflurane, and their heads were fixed using a KOPF stereotaxic assembly (David Kopf Instruments, Tujunga, CA, USA). A midline incision was made over the scalp, and a burr hole was made on the skull with a drill (bregma: 0.5 mm, lateral: 3.0 mm). The 29-Ga thermocouple was then inserted through the burr hole into the caudate-putamen, and fixed in place using dental cement.
Blood pressure and heartbeat rates were continuously monitored through a left femoral arterial catheterization, using a Power Lab system (AD Instruments, Colorado Springs, CO, USA). At indicated time points, arterial blood was collected from the femoral catheterization and analyzed for blood gas and electrolytes using an iSTAT Portable Clinical Analyzer (Abbott Point-of-Care, East Windsor, NJ, USA) combined with CG8 + cartridges (Stender et al, 2007).
Preparation and Administration of Adenosine 5'-Monophosphate and Compound C
Adenosine 5'-monophosphate was purchased from Sigma (St Louis, MO, USA) and dissolved in sterile distilled water. Freshly prepared AMP solution was injected intraperitoneally at dosages of 2.5, 4, or 5 mmol/kg body weight for the hypothermic studies. In AMP-treated ischemia groups, 4 mmol/kg of AMP was injected intraperitoneally 30 mins after the onset of cerebral ischemia, which is 90 mins before reperfusion. Compound C, also known as dorsomorphin or 6-[4-(2-Piperidin-1-yl-ethoxy)-phenyl)]-3-pyridin-4-yl-pyrrazolo [1,5-a]-pyrimidine, is a selective inhibitor of AMP-activated protein kinase (AMPK). Compound C was purchased from Calbiochem (Gibbstown, NJ, USA), and dissolved in dimethyl sulfoxide (DMSO) to make a stock solution. A work solution was prepared by diluting the stock solution with normal saline, and injected intraperitoneally at doses of 5 and 10 mg/kg body weight.
Induction of Mild Hypothermia in the Brain
To determine whether brain hypothermia protects the brain from ischemic injury, we induced mild brain hypothermia in isoflurane-anesthetized rats by applying an icepack directly over the head and neck, while the CBT was maintained within the physiologic range using temperature-regulated heating pad and lamp as described earlier (Luo et al, 2007). Under continuing brain temperature monitoring, brain hypothermia was initiated 30 mins before postischemic reperfusion and maintained at 33 ± 0.5°C for 3h after reperfusion (Luo et al, 2007). The rats in the normothermic control group were treated similarly, except that the brain temperature was maintained within the physiologic range without an icepack.
Rat Model of Transient Focal Cerebral Ischemia
Transient focal cerebral ischemia was induced by middle cerebral artery occlusion (MCAO) in rats as described earlier with slight modifications (Karibe et al, 1994; Luo et al, 2007; Zhang et al, 1998). In brief, male rats were anesthetized with 1.5% to 2% isoflurane in a mixture of 30% O2:70% N2O for either a shorter duration with a facemask or for a longer duration with trachea intubation and mechanical ventilation. For the shorter duration, rats were anesthetized only during the surgical operation; this applied to behavioral study and ischemic outcome studies. For the longer duration, rats were anesthetized throughout the entire procedure, and their CBT was adjusted according to experimental design; this applied to the studies of physiologic parameter monitoring. The left external, internal, and common carotid arteries were exposed through a midline neck incision. After coagulating and cutting of the branches of the external carotid arteries, a 3-0 monofilament nylon suture with a blunted tip was inserted into the lumen of the external carotid artery, advanced into the internal carotid artery, and then into the origin of the middle cerebral artery. The rats underwent an MCAO for 2 h followed by reperfusion for the indicated duration.
A change in regional cerebral blood flow (rCBF) was evaluated in a separate group using laser-Doppler flowmetry. Regional cerebral blood flow was measured at three locations namely, ischemic core (bregma: 1.0 mm, lateral: 3.0mm), penumbra (bregma: −3.0 mm, lateral: 4.0 mm), and contralateral hemisphere (bregma: 1.0 mm, lateral: 3.0mm). Four sequential measurements were performed on each rat at 5 mins before MCAO, 20 mins after the onset of MCAO (10 mins before intraperitoneal injection), 60 mins after the onset of MCAO (30 mins after intraperitoneal injection), and 30 mins after the onset of reperfusion.
Evaluation of Ischemic Outcomes
Ischemic outcomes in rats were assessed using neurologic dysfunction, infarct volume, hemorrhagic transformation (HT), and rates of death and seizure. For this purpose, the rats were randomly divided into three groups, such as vehicle-treated, AMP-treated, and external cooling (ice bag)-induced hypothermia, with 7 to 10 animals per group. Their neurologic dysfunction was evaluated at 24 h after ischemia and scored on a 0 to 4 scale, with 0 being the least and 4 the worst. To quantify infarct volumes induced by focal cerebral ischemia, rats were killed at 24 h after MCAO, and their brains were removed and sliced into 7 coronal sections of 2 mm thickness. The sections were immediately incubated with 2% of 2, 3, 5-triphenyltetrazolium chloride (TTC). The infarct size in each section was measured using the MCID image analysis system (Imaging Research Inc., St Catharines, ON, Canada), and was then integrated into infarct volume. Hemorrhagic transformation was also evaluated on TTC-stained brain sections, which appeared as punctated dark dots on the white background of the TTC-stained brain sections.
Measurement of Serum Insulin Concentration
To prepare serum, rats from the vehicle- and AMP-treated groups (n = 4 per group) were anesthetized and their blood was collected through a left femoral arterial catheterization every 90 mins. The whole blood samples were allowed to clog at room temperature for 60 mins and then centrifuged for 15 mins at 4°C. The serum were collected and stored in a −80°C freezer until use. The serum insulin concentration was measured using a Rat Insulin ELISA Kit from Millipore (Billerica, MA, USA) according to the manufacturer's protocols.
Western Blot Analysis
Western blots were performed using the standard method described earlier (Luo et al, 2007). Rats were killed at 1, 4, and 8 h after MCAO, or 8 h after sham operation (n = 3 to n = 4 per experimental condition). In the AMP-treated groups, 4 mmol/kg of AMP was injected intraperitoneally 30 mins after the onset of MCAO. The cortical tissues from both the ipsilateral and the contralateral hemispheres were collected, frozen with dry ice, and stored in a −80°C freezer until use. After thawing, the brain tissues were homogenized in lysis buffer containing 1% of protease inhibitor cocktail. The suspension was then sonicated and centrifuged. The total protein extracts were separated using 4% to 15% gradient Ready Gels (Bio-Rad, Hercules, CA, USA), and subjected to western blot analysis. Blots were probed with antibodies recognizing total acetyl-CoA carboxylase (ACC) and phosphorylated ACC at Ser79, and total AMPK± (t-AMPK±) and phosphorylated AMPK± (p-AMPK±) at Thr172. The primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). The optical density of bands was measured with the assistance of MCID Image Analysis Software (Imaging Research Inc., Cambridge, UK). For gel analysis, the optical density of each band was first normalized with their corresponding β-actin band, then compared with that of a sham-operated animal on the same gel.
Statistical Analysis
Data on temperature, infarct volume, heartbeat, blood pressure, insulin, blood glucose, and electrolytes are presented as mean ± s.d., and analyzed using ANOVA (analysis of variance) followed by post hoc Scheffe's tests. Data on animal death rate, seizure rate, and HT rate were presented as a ratio and analyzed using χ2-test. Data on the optical density of western blot bands were presented as mean ± s.e., and analyzed using ANOVA followed by post hoc Scheffe's tests. Statistically, significance was considered when the P-value was < 0.05.
Results
Adenosine 5'-Monophosphate Induces Hypothermia in Rats
To determine whether AMP induces hypothermia in rats, the AMP solution was injected intraperitoneally. The doses of AMP ranged from 2.5 to 5 mmol/kg, based on earlier studies with mice (Swoap et al, 2007; Zhang et al, 2006). As shown in Figure 1A, 2.5 mmol/kg of AMP decreased the CBT to only 34.7 ± 0.6°C, which is not in the mild hypothermia range. We then increased the AMP dosage to 4 mmol/kg, and observed that CBT was decreased to below 34°C at 100 mins after AMP injection. This mild hypothermia remained for 2.5 h, with the lowest CBT at 33.1 ± 0.7°C. Adenosine 5'-monophosphate at 5 mmol/kg body weight decreased CBT to below 34°C even faster, and maintained hypothermia longer; however, one rat died at 5h after the injection. During the hypothermic periods, the rats were awake without obvious shivering, but were less active. Taken together, these data indicate that AMP induces hypothermia in a dose-dependent manner, and 4 mmol/kg of AMP is an appropriate dose to induce mild hypothermia of 32°C to 34°C in rats (Maier et al, 1998; Miyazawa et al, 2003). Therefore, we used AMP at a dose of 4 mmol/kg in our subsequent experiments.
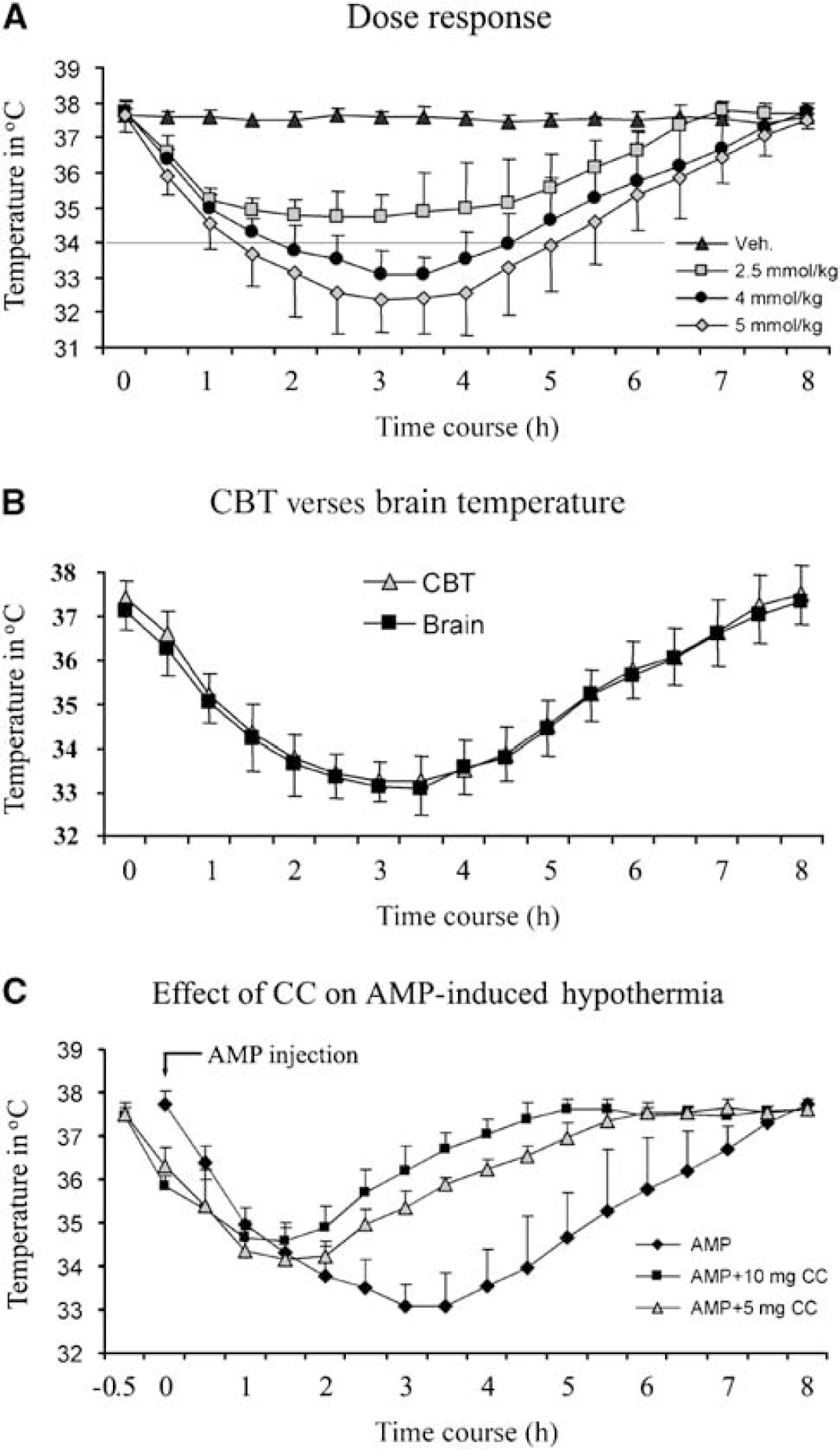
AMP induces hypothermia in rats. (
We next investigated the correlation between brain temperature and CBT. To do this, the rats were anesthetized with isoflurane and intubated with mechanical ventilation; their brain temperatures and CBT were measured simultaneously. As shown in Figure 1B, the brain temperature was 0.1°C to 0.2°C lower than CBT before AMP injection. As CBT decreased gradually after AMP injection, the brain temperature dropped concurrently, indicating that it is in parallel with CBT.
In their report, Zhang et al, 2006 proposed a possible role for AMPK in AMP-induced hypothermia. To determine the actual role of AMPK in hypothermia, we performed an AMPK inhibition study with Compound C, a specific AMPK inhibitor. Compound C dissolved in DMSO/saline was injected intraperitoneally 30 mins before the injection of AMP. When 5mg/kg of Compound C was injected, the duration of hypothermia was shortened, and the lowest temperature was higher compared with the AMP-only group (Figure 1C). When Compound C was administered at a dose of 10 mg/kg, which was only half of the optimal inhibitive dose (Li et al, 2007), the hypothermic effect of AMP was further compromised (Figure 1C). To avoid the possibility that DMSO has any effect on AMP-induced hypothermia, we carried out two control experiments using DMSO. In the first group, the rats were treated with DMSO dissolved in normal saline, and this DMSO treatment did not induce hypothermia in them. In the second group, the rats were treated with DMSO followed by 5'-AMP infusion; and we found that DMSO did not have significant influence on 5'-AMP-induced hypothermia (data not shown). Taken together, these data indicate that AMPK may play an important role in AMP-induced hypothermia in rats.
Adenosine 5'-Monophosphate Exacerbates Ischemic Brain Injury in Rats
We next aimed at determining whether AMP-induced hypothermia provides neuroprotection against ischemic injury induced by transient focal cerebral ischemia in rats. The first experiment that we performed was to confirm that brain hypothermia does protect the brain from ischemic injury induced by a 2-h MCAO in rats. We induced mild hypothermia in rat brains using an icepack as described earlier (Luo et al, 2007), starting 30 mins before postischemic reperfusion and maintaining at 33 ± 0.5°C for 3h after reperfusion. As shown in the left panels of Figures 2A and 2B, brain hypothermia reduced the infarct volume to 106.3 ± 45.3 mm3 from 256.4 ± 31.6 mm3 in the normothermic group. The difference in physiologic parameters between the hypothermic and normothermic groups was not significant before, during, and after the MCAO (data not shown).
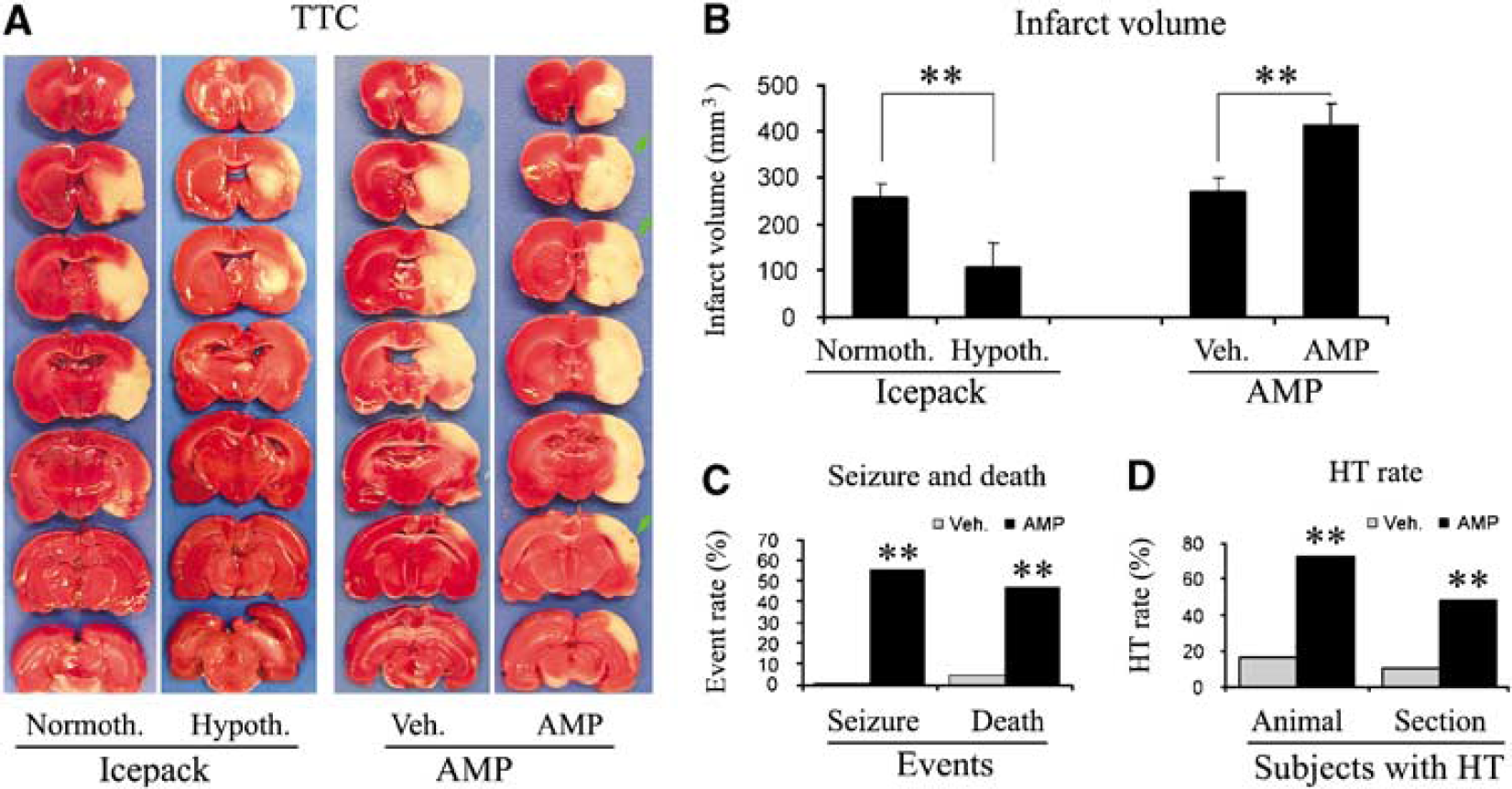
The adverse effect of AMP on ischemic outcomes in rats. (
To determine whether AMP-induced hypothermia provides a similar neuroprotection against ischemic injury in rats, we induced ischemia in rats by a 2-h MCAO. Adenosine 5'-monophosphate of 4 mmol/kg was injected intraperitoneally at 30 mins after the onset of ischemia. In this way, mild hypothermia could be achieved at the onset of reperfusion and last for 2 to 3h. Unexpectedly, AMP exacerbated the ischemic outcomes. This was first indicated by an increased rate of seizure after MCAO in the AMP-treated group, which occurred in ~55% of rats versus 0% in the vehicle-treated group (Figure 2C), and usually occurred at 8h after ischemia or later. Increased mortality was also observed in the AMP-treated group (Figure 2C), and doomed rats usually died of coma or seizure. The TTC stain confirmed the adverse effect of AMP, showing that AMP increased the infarct volume by 53.5%, from 269.2 ± 33.9mm3 in the vehicle group to 413.2 ± 48.4 mm3 in the AMP group (Figures 2A and 2B). Furthermore, the AMP-treated group showed an increased occurrence of HT (Figure 2A), which appeared as punctated bleeding in the infarct areas. The percentages of both HT sections and HT animals were significantly increased in the AMP-treated group (Figure 2D).
Adenosine 5'-Monophosphate Causes Cardiovascular Depression and Cerebral Hypoperfusion in Rats
The mechanisms that underlie the adverse effects of AMP on ischemic outcomes must be fatal or multiple factors, because they are able to overturn the robust neuroprotective effects of hypothermia. Therefore, we aimed at determining the detrimental factors of AMP. As a result of decreased energy demand, hypothermia is characterized by depressed cardiovascular function (Kiyatkin and Brown, 2005; Yenari et al, 2008), and hypotension per se can decrease cerebral blood flow and exacerbate ischemic injury (Zhu and Auer, 1995). Recently, AMP was reported to be able to cause bradycardia in mice (Swoap et al, 2007); hence, we hypothesized that AMP may cause cardiovascular depression, which in turn contributes to the adverse effect of AMP on brain ischemia.
To test our hypothesis, we monitored arterial blood pressure and heartbeat rate in rats by femoral catheterization. For the monitoring, the rats were anesthetized with isoflurane and mechanically ventilated with tracheal intubation. The CBT was maintained at the physiologic range in the vehicle group, and adjusted in accordance with the temperature curve in the AMP-treated group (Figure 1A). As shown in Figure 3, AMP did decrease blood pressure, and its suppressive effect on blood pressure was rapid and dramatic. The blood pressure decreased to ~80 mmHg when the injection was completed; it quickly decreased to ~60 mmHg at 1 min after AMP injection and to 40 mmHg at 2 mins (Figure 3A). This hypotension continued for the next 3h, and then recovered slowly (Figure 3B). Adenosine 5'-monophosphate also reduced the heartbeat rate (Figure 3C). The decrease in the heartbeat rate occurred in a relatively milder manner compared with that in blood pressure, with the lowest rate of 248 ± 47 b.p.m. at 2h after AMP injection.
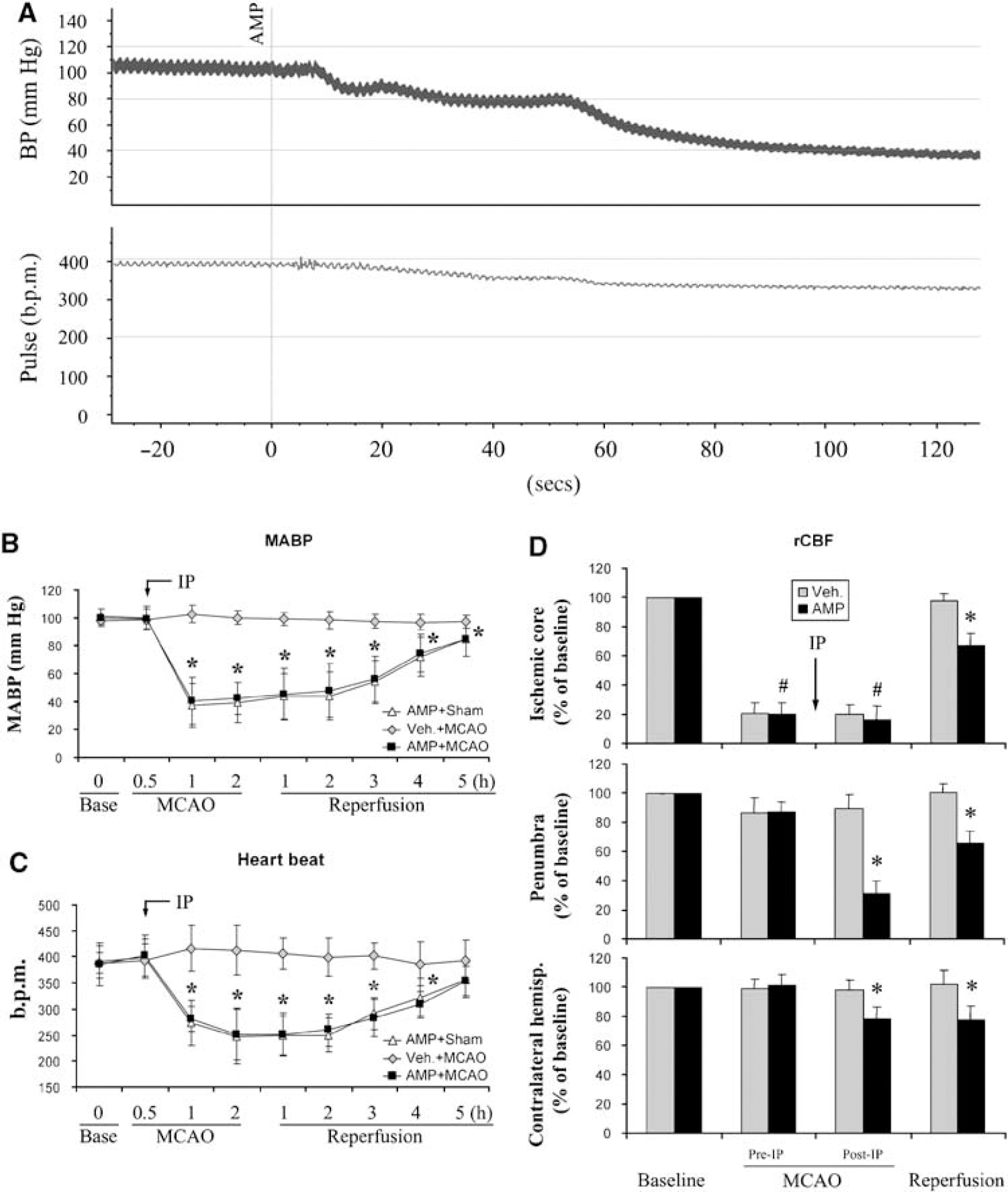
AMP causes cardiovascular depression and cerebral hypoperfusion in rats. (
To investigate whether the AMP-induced cardiovascular depression impairs cerebral blood flow, we used laser-Doppler flowmetry to measure rCBF, which is presented as a percentage of the baseline value. In the ischemic core, AMP did not further decrease rCBF during MCAO; however, it allowed only a partial restoration of rCBF during reperfusion (Figure 3D, upper panel). In the penumbra, a fatal reduction in rCBF was noticed in the AMP-treated group, and this change was observed during both ischemia and reperfusion (Figure 3D, middle panel). In the contralateral cortex, a slight decrease in rCBF was also detected in the AMP-treated rats (Figure 3D, lower panel). Taken together, these data suggest that AMP induces cardiovascular depression in rats, which consequently impairs rCBF and results in postischemic hypoperfusion.
Although cardiovascular depression plays an important role in worsening ischemic injury, it may not be the only detrimental factor in the adverse effect of AMP. Adenosine, a derivative of AMP, causes a similar cardiovascular depression (Kassell et al, 1983; Stange et al, 1989) and hypothermia (Swoap et al, 2007), but provides neuroprotection against cerebral ischemia (Sweeney, 1997). Therefore, we investigated whether AMP has other injurious factors.
Adenosine 5'-Monophosphate Induces Hyperglycemia, Metabolic Acidosis, and Hypocalcemia
Hyperglycemia is a notorious factor that intensifies ischemic injury in the brain (Li and Siesjo, 1997; Prado et al, 1988). Large infarct, HT, and increased rates of seizure and death are features of the hyperglycemic effect on brain ischemia (Li and Siesjo, 1997). We observed that the adverse effect of AMP on ischemia showed those characteristics, and therefore, hypothesized that hyperglycemia may be a detrimental aspect of AMP in brain ischemia. To test this, we measured blood glucose levels in nonfasted rats, along with electrolytes and arterial blood gases.
Blood samples were collected by femoral catheterization and then analyzed using an iSTAT Portable Clinical Analyzer. In the vehicle-treated group, blood glucose concentration remained stable for the first 2 h during monitoring, at an average of 243 ± 29 mg/dL, and slightly decreased thereafter (Figure 4A). In contrast, glucose levels in the AMP-treated groups dramatically increased; they reached 422.5 ± 39 mg/dL 30 mins after the injection and peaked at 619.5 ± 72 mg/dL 90 mins after the injection (Figure 4A), indicating severe hyperglycemia. Blood glucose levels then diminished gradually. Between the two AMP-treated groups, MCAO did not significantly alter the blood glucose concentration.
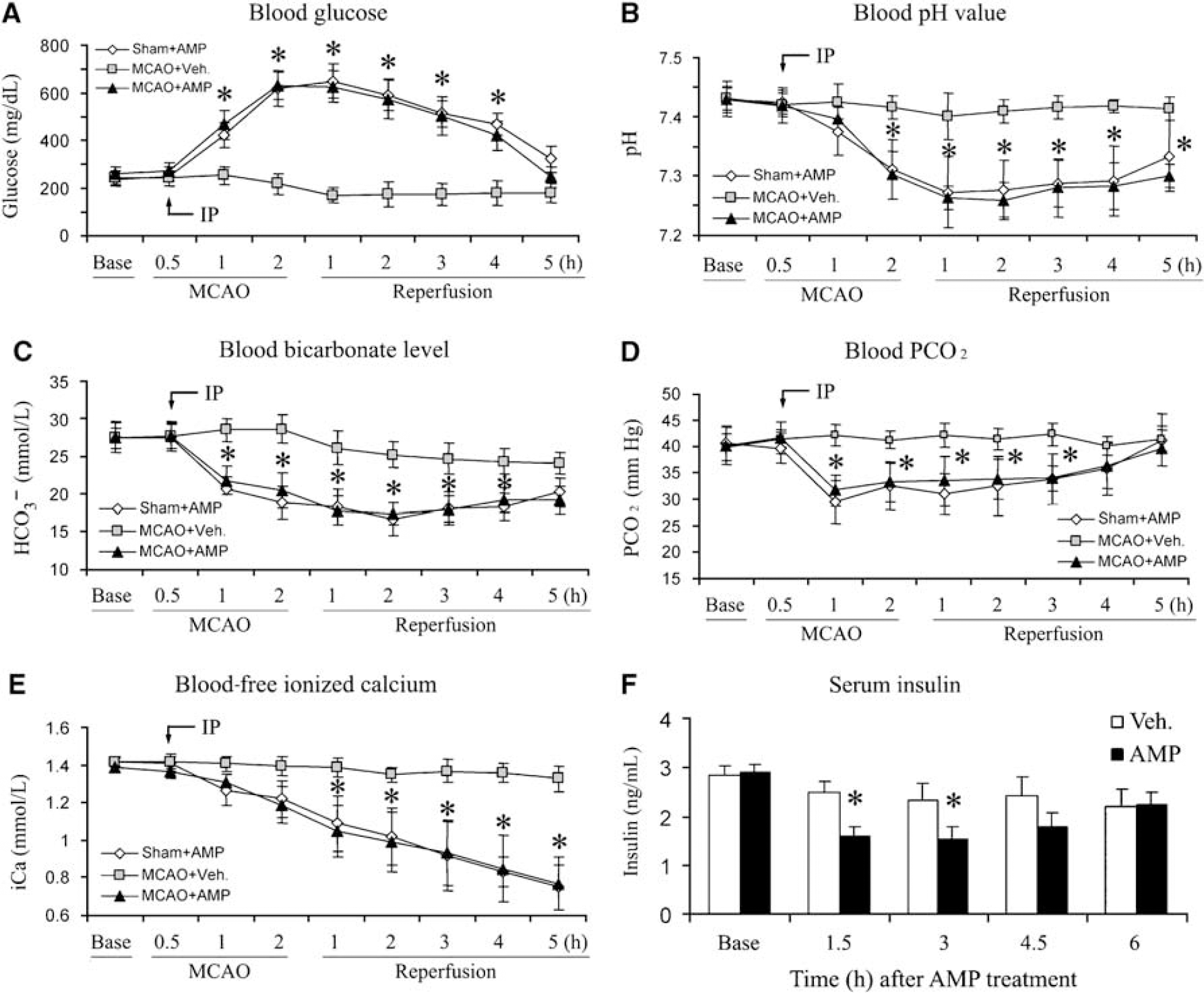
AMP causes hyperglycemia and metabolic acidosis. (
Along with the hyperglycemia, blood pH value decreased sharply after AMP injection (Figure 4B). It reached 7.31 ± 0.05 at 90 mins after AMP injection, and bottomed at 2.5 h with a pH of 7.26 ± 0.05. Accompanying the decrease in pH, the blood bicarbonate level was comparably decreased (Figure 4C), and a parallel reduction in blood PCO2 also occurred (Figure 4D), indicating that AMP induces severe metabolic acidosis.
Blood sodium concentration remained stable in all the three groups after AMP injection or MCAO (data not shown). However, a moderate hyperkalemia slowly developed ~4h after AMP administration, reaching 6.6 ± 0.9 mmol/L at 7h after the injection (data not shown). Unexpectedly, the blood concentration of free ionized calcium began decreasing after AMP injection; it became significant at 90 mins after the injection, and continued getting worse as time progressed (Figure 4E). This severe hypocalcemia may be a contributory factor to the increased rate of seizure and animal death after MCAO. Blood potassium and calcium concentrations returned to near normal 24 h after the injection, which were measured when the rats were killed.
The dominant cause of hyperglycemia and metabolic acidosis is the absence or insufficiency of insulin, and it has earlier been reported that the activation of AMPK in pancreatic â-cells decreased the production of insulin (Cai et al, 2008; Tsuboi et al, 2003). Therefore, we hypothesized that the administration of AMP may result in a decrease in serum insulin levels in rats. To test this hypothesis, we measured serum insulin concentration at various time points after the administration of AMP at the dose of 4 mmol/kg. As shown in Figure 4F, the insulin levels in the AMP-treated group were significantly lower than that in the vehicle-treated group at 1.5 and 3h, and then gradually recovered. Taken together, these data suggest that the administration of AMP decreases the serum insulin level, which in turn results in hyperglycemia and acidosis.
Effects of Fasting on Physiologic Parameters and Ischemic Infarct
All the experiments up to this point were carried out using nonfasted rats. To reduce the adverse effects of hyperglycemia, we decided to study the effects of fasting on physiologic factors and ischemic outcomes in rats. For this purpose, the rats were deprived of food overnight, with free access to water. The CBT in fasted rats was 0.8 ± 0.2°C, which was lower than that in nonfasted rats (Figure 5A). Compared with that in nonfasted rats, a deeper hypothermia was induced in fasted rats after the administration of AMP, with a longer duration of mild hypothermia (Figure 5A).
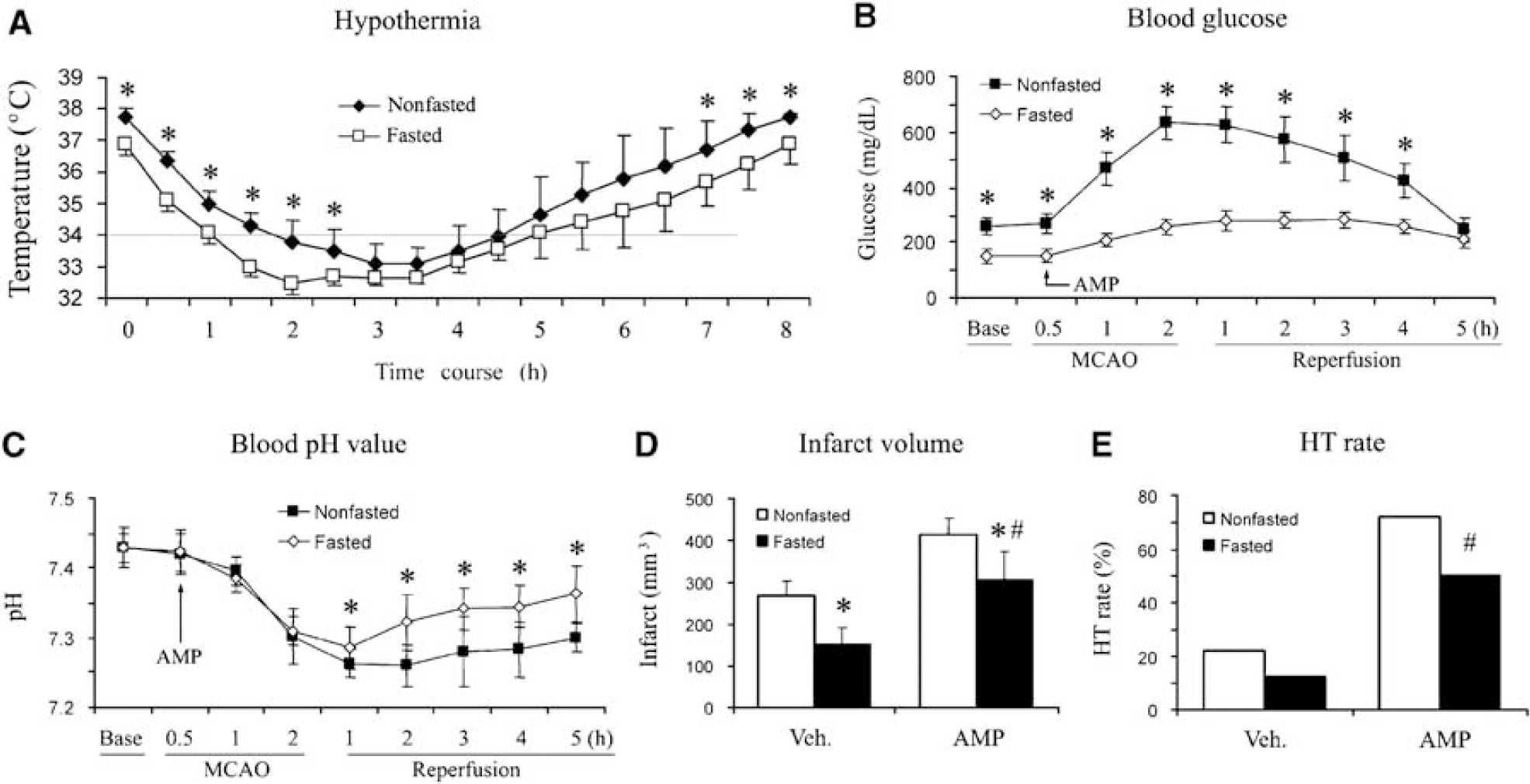
The effect of fasting on hypothermia and infarct in rats. (
In fasted rats, the blood glucose concentration was 149 ± 27mg/dL, which was significantly lower than 243 ± 29 mg/dL in nonfasted rats (Figure 5B). In contrast to the striking hyperglycemic response in nonfasted rats, AMP caused only moderate hyperglycemia in fasted rats, with a maximal concentration of 281.5 ± 28 mg/dL (Figure 5B). The metabolic acidosis induced by AMP was also improved in fasted rats (Figure 5C).
Consistent with earlier reports (Lin et al, 1997; Yip et al, 1991), in fasted rats after focal ischemia, we detected a smaller infarct volume, 152.2 ± 40 mm3, which represents a 47% reduction compared with that in nonfasted rats (Figure 5D). After AMP treatment, the infarct volume in fasted rats was also decreased compared with that in the nonfasted, although the harmful effect of AMP remained (Figure 5D). Similarly, fasting partially reduced HT rates in the AMP-treated groups after MCAO. Taken together, our data indicate that fasting enhances hypothermia, attenuates the hyperglycemia and metabolic acidosis induced by AMP, and reduces infarct volume after MCAO, but cannot completely prevent the detrimental effect of AMP on ischemic consequence.
Adenosine 5'-Monophosphate-Activated Protein Kinase in Rat Brain After Ischemia
The AMPK acts as an energy sensor and switch, and its activity is upregulated through the phosphorylation of its catalytic ±-subunit at Thr172, as well as an increased ratio of AMP-ATP (Young, 2008). An unfavorable effect of AMPK on brain ischemia has been reported recently (Li et al, 2007; McCullough et al, 2005). As the administration of exogenous AMP in rats will increase the ratio of AMP-ATP, we therefore, assume that AMPK activity in the rat brain would be enhanced after AMP treatment and subsequently contribute to the adverse effect of AMP on ischemic outcome. To ascertain this, we collected brain tissues at serial time points after ischemia, and then performed western blots to detect the levels of p-AMPK± and t-AMPK±.
In the vehicle-treated group, the levels of p-AMPK± and t-AMPK± in the ischemic cortex were not significantly changed during the first 4h after ischemia, but slightly decreased at 8 h after ischemia (Figures 6A and 6B). Surprisingly, we did not observe any increase in the levels of p-AMPK± in the ischemic cortex after AMP treatment (Figures 6A and 6B); instead, the p-AMPK± level was sharply decreased in the AMP-treated groups. To explore the underlying reason for the p-AMPK decrease, we measured the t-AMPK± expression. The result showed a similar decrease in t-AMPK± levels in the ischemic cortex of the AMP-treated rats (Figures 6A and 6B), indicating a rapid protein degradation in the AMP-treated groups. To confirm this finding, we measured the phosphorylation of one specific substrate of AMPK, acetyl-CoA carboxylase (ACC), at Ser79. As shown in Figure 6A, the levels of phosphorylated ACC in the AMP-treated groups were much lower than those in the vehicle-treated groups, verifying a rapid protein degradation and tissue damage in the ischemic core of the AMP-treated rats.
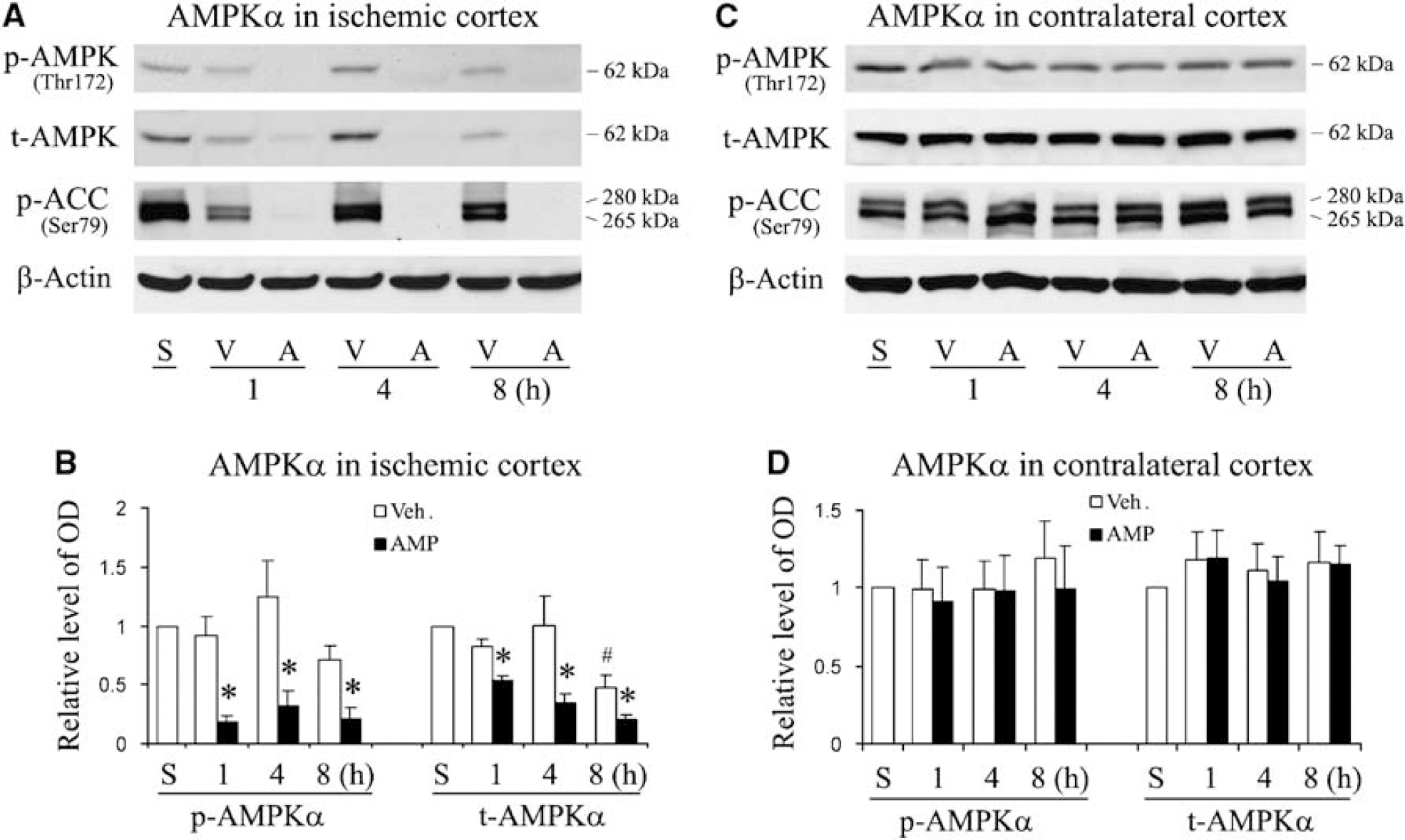
AMPK± in rat brain after MCAO. (
To investigate whether exogenous AMP has any effect on the AMPK± activity of the intact brain, we performed western blots with the cortex tissue from contralateral hemispheres of rat brains. As shown in Figures 6C and 6D, no significant changes in the levels of p-AMPK± and t-AMPK± were detected from the contralateral cortex after brain ischemia, indicating that exogenous AMP did not cause an alteration in AMPK± activity in the intact brain. Taken together, these findings suggest that brain AMPK± was degraded at an early stage in the ipsilateral cortex after focal brain ischemia in the AMP-treated group, and therefore, that AMPK± may not make a significant contribution to the overall results of ischemic injury in our experiments.
Discussion
Hypothermia is an established approach that protects the brain from acute ischemic brain damage, and a majority of CBT-lowering methods have so far been effective in this regard (Yenari et al, 2008). However, the results of this study indicate that whether a compound is able to protect the brain through hypothermia depends on its hypothermic mechanisms and side effects.
The precise mechanisms underlying the induced hypothermia are unclear, but they consist of a decrease in heat production and an increase in heat dissipation. Adenosine 5'-monophosphate causes cardiovascular depression, which is manifested by inhibitory signs of sympathetic nervous system, such as bradycardia and hypotension (Figure 3). This cardiovascular depression may contribute to the hypothermic effect of AMP (Swoap et al, 2007), because the hypotension limits glucose and oxygen delivery to tissues, and vasodilation increases heat loss. However, it is unclear how AMP causes cardiovascular depression. Adenosine, a derivative of AMP, causes cardiovascular responses similar to those of AMP (Stange et al, 1989; Swoap et al, 2007). Therefore, it is possible that AMP is dephosphorylated to adenosine by ecto-5'-nucleotidase located at endothelial cells, and that adenosine then activates adenosine receptors in the heart and blood vessels (Stange et al, 1989; Swoap et al, 2007), leading to cardiovascular depression. It is also possible that AMP will directly activate AMPK in endothelial cells, leading to increased nitrous oxide production and vessel dilation (Suzuki et al, 2008; Xu et al, 2008).
Glucose is the major fuel for the production of ATP and heat, so it is reasonable to hypothesize that hypothermia could be induced by a decreased usage of glucose, especially in the major heat-producing organs, such as the muscle and liver. Recently, three compounds have been reported to be able to induce hypothermia in animals namely, hydrogen sulfide (Blackstone et al, 2005), 2-deoxy-
In terms of severe hyperglycemia and metabolic acidosis, we observed a similarity between the following two conditions: the rats with AMP administration and those with type I diabetes and ketoacidosis. Therefore, we hypothesize that an acute lack of insulin could be the reason for AMP-induced hyperglycemia and hypothermia. Earlier it has been reported that the activation of AMPK in the pancreatic â-cells inhibits their insulin secretion (Tsuboi et al, 2003; Wang et al, 2007), and even causes their apoptosis (Cai et al, 2008). These events will inevitably result in decreased insulin concentration in blood as shown in Figure 4F, which may consequently cause hypothermia and hyperglycemia with the mechanisms discussed below.
The actual effect of insulin on glucose transport depends on the type of glucose transporter (GLUT). There are two major types of glucose transporters namely, insulin-independent and insulin-dependent (Dugani and Klip, 2005; Simpson et al, 2007). Insulin-independent glucose transporters include GLUT1 and GLUT3, with the former expressed in microvascular endothelial cells in the brain and glia, and the latter in the neuron. Insulin-dependent glucose transporter mainly includes GLUT4, which is broadly expressed in the liver, muscle, and adipocytes. Thus, when the insulin level decreases, blood glucose cannot readily enter into cells in the liver and muscle through GLUT4, resulting in hyperglycemia. As a result, the muscle and liver would have a limited supply of glucose, leading to a decrease in heat production and thus hypothermia. Simultaneously, the glucose built-up in blood would flood into the brain through insulin-independent GLUT1 and GLUT3.
Hyperglycemia is an established adverse factor in ischemic injury in the brain, and its threshold for increasing ischemic injury is ~12 mmol/L, which is 216 mg/dL (Li and Siesjo, 1997). The blood glucose concentration was much higher than the threshold concentration after AMP administration; therefore, it was not surprising that AMP exacerbated ischemic damage. One signature effect of hyperglycemia on brain ischemia is an increased seizure rate; the reason for this phenomenon is not clear, although free radicals have been proposed as the mediators (Li and Siesjo, 1997). As an interesting novel finding, we found that a sustained hypocalcemia accompanies hyperglycemia and acidosis after AMP treatment, and the hypocalcemia may therefore be the reason for the increased seizure rate after stroke. The mechanisms for the hypocalcemia are not clear, although parathyroid hormone suppression is suggested (D'Erasmo et al, 1999). In humans, it was reported that hyperglycemia induced by oral glucose load caused a slight decrease in serum calcium (1.28 to 1.27 mmol/L) and an increase in urinary calcium discharge. Those were in parallel with a decrease in parathyroid hormone level (D'Erasmo et al, 1999). However, the hypocalcemia we observed is much more severe than that described in an earlier report, and the reasons remain elucidated.
Hemorrhagic transformation is another feather of the adverse effect of hyperglycemia on cerebral ischemia and reperfusion. The rate of HT in hyperglycemic cats was five times more than that in normoglycemic animals after MCAO and reperfusion (de Courten-Myers et al, 1992), and hyperglycemia is associated with a consistent HT after MCAO and reperfusion in rats (Qin et al, 2007). Therefore, it is quite likely that AMP-induced hyperglycemia might have contributed to the increased rate of HT after ischemia in our study. Evidence shows that matrix metalloproteinase-9 plays an important role in the dysfunction of the blood-brain barrier after stroke, and hyperglycemia increases oxidative stress and matrix metalloproteinase-9 activity (Kamada et al, 2007; Tsuji et al, 2005; Yang et al, 2007), implying the potential mechanism on the hyperglycemia-induced HT. However, additional mechanisms must be involved in AMP-induced HT after ischemia; as in fasted rats, for which the hyperglycemic effect of AMP was diminished, the rate of HT was lower than in nonfasted rats but was still significantly higher than in fasted rats without AMP treatment.
Fasting in experimental animals reduces their blood glucose level and decreases infarct volumes after brain ischemia. For example, fasting rats for 24 h decreased their infarct volume by 50% to 75% after MCAO (Lin et al, 1997; Yip et al, 1991). Our result was in line with these reports, showing a 47% reduction in the infarct volume in rats with overnight fasting. Adenosine 5'-monophosphate treatment in fasted rats led to an improved hypothermic effect and blunted hyperglycemia compared with that in nonfasted rats; however, it still resulted in a larger infarct than in the vehicle-treated fasted rats, confirming the detrimental effect of AMP on ischemic outcome.
The AMPK functions as a metabolic master switch in the regulation of energy homeostasis, including glucose transport and metabolism, as well as biogenesis of protein and lipid. AMPK activity is regulated by the phosphorylation of its catalytic ±-subunit at Thr172, as well as the ratio of AMP-ATP (Young, 2008). The activated AMPK will phosphorylate ACC to inhibit fatty acid synthesis; it also phosphorylates mammalian target of rapamycin to inhibit protein synthesis. The role of AMPK in cerebral ischemia is still controversial. Using AMPK inhibitors and mice deficient in AMPK±, one group showed a detrimental role of AMPK in brain ischemia (Li et al, 2007; McCullough et al, 2005). They also showed that an AMPK activator, 5-aminoimidazole-4-carboxamide ribonucleoside, worsened the ischemic injury in mice (McCullough et al, 2005). In contrast, 5-aminoimidazole-4-carboxamide ribonucleoside was shown to be able to reduce the ischemic damage in gerbils (Clough-Helfman and Phillis, 1990) and to protect hippocampal neuronal cultures against injuries induced by glucose deprivation, chemical hypoxia, and glutamate exposure (Culmsee et al, 2001). Recently, AMPK was shown to phosphorylate and activate neuronal gamma-aminobutyric acid type B (GABAB) receptors, and to enhance neuronal survival after ischemic injury (Kuramoto et al, 2007). However, in this study, we were prevented from clarifying the role of AMPK in ischemic injury in vivo by the early death of the brain tissue caused by AMP. In a further effort to understand the role of AMP in ischemic neuronal injury, we performed in vitro ischemic experiments using primary neuronal culture and oxygen-glucose deprivation. Adenosine 5'-monophosphate treatment decreased lactate dehydrogenase (LDH) release by 10% to 15% compared with the vehicle group (data not shown); however, the difference was not significant, suggesting that AMPK may not play a key role in neuronal ischemic death.
In Figure 7, we summarized the AMP-stimulated hypothermia, hypotension, and hyperglycemia (ASH) and its influence on ischemic outcomes. Systemic administration of exogenous AMP increases AMP concentration in blood, which then activates AMPK in the pancreas and cardiovascular system. Pancreatic AMPK activation reduces insulin production and release, limits glucose entry into cells outside the central nervous system (CNS) and therefore results in hyperglycemia. Cardiovascular AMPK activation may cause cardiovascular depression, leading to hypotension and hypoperfusion. Adenosine 5'-monophosphate-induced hypothermia may result from a reduced delivery and transport of glucose. Hypothermia may offer the brain protection against ischemic injury; however, this neuroprotective effect is overwhelmed by the adverse effects of hyperglycemia and cerebral hypoperfusion. The role of AMPK activation in cerebral ischemia remains to be clarified.
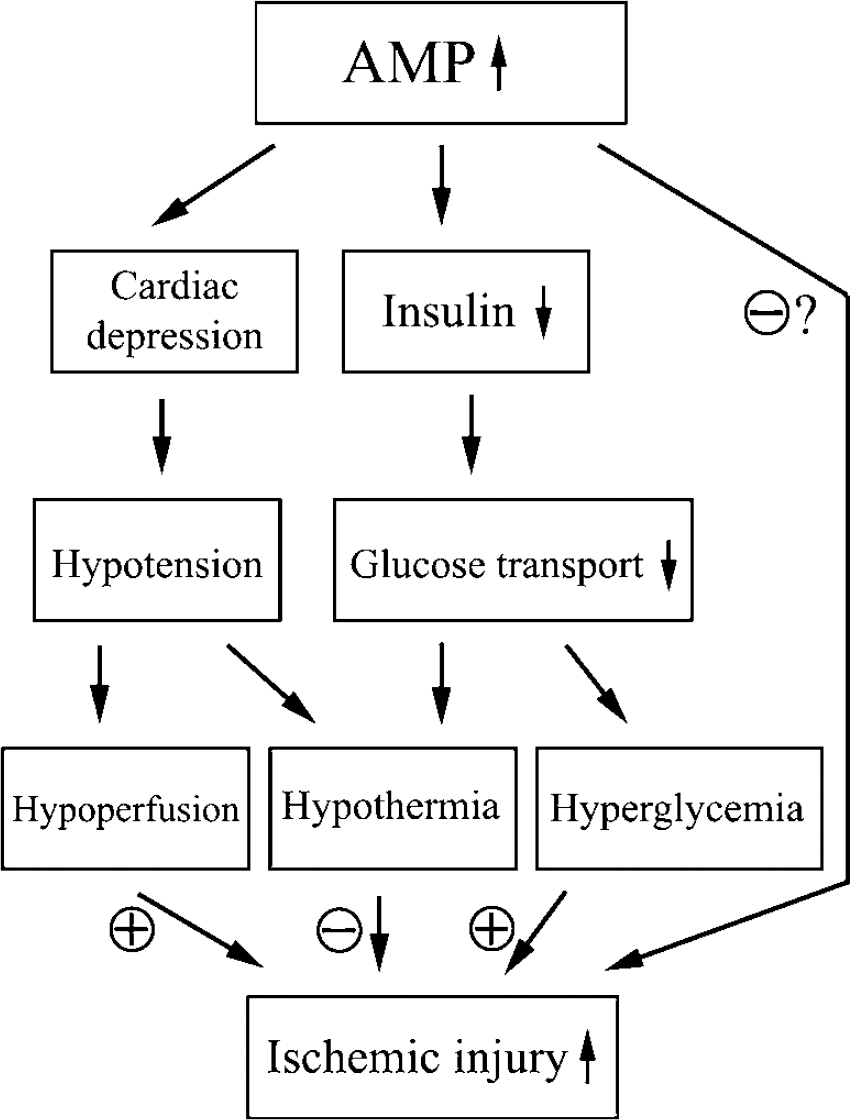
Diagram showing the diverse effects of AMP on brain ischemia.
The significance of this study is multiple. To begin with, we show that AMP successfully induces hypothermia and torpor-like states in rats in a dose-dependent manner. In addition, AMP may be a tool for creating an animal model of acute short-term type I diabetes and diabetic ketoacidosis. Furthermore, we show that hypocalcemia is associated with hyperglycemia and acidosis and therefore, might be the causal factor in increased seizure rate in hyperglycemic stroke. Unfortunately, AMP is unsuitable for inducing hypothermia to protect the brain from ischemia, because it causes hyperglycemia and hypotension, which badly exacerbates ischemic brain injury. Finally, our findings may imply that when a compound is systemically administered, or when the animal used is manipulated genetically, special attention should be paid to the systemic responses, especially the blood sugar, blood gases, and vital signs.