
Abstract
Pharmaceutical profiling is the characterization of the physicochemical and metabolic properties of compounds during drug discovery. The data alerts project teams to compound liabilities, helps to diagnose complex in vivo processes such as bioavailability, helps to better plan and interpret discovery experiments, and provides feedback for synthetic chemical modifications to the structure that are designed to improve properties. Automation plays an important role in obtaining diverse sets of property data for thousands of compounds per year with short turnaround times that match discovery workflow. Key elements of automation in profiling include liquid handling, high capacity detection, and data processing and reporting. Key information is obtained from high throughput assays for integrity, permeability, solubility, stability, cytochrome P450 inhibition, lipophilicity, and pKa. Automated assays for these properties are discussed.
Introduction
Pivotal studies in the late 1980s alerted pharmaceutical researchers to the main causes of failure of drug candidates during development. While percentages may differ between studies, two prominent causes of failure were reported as poor biopharmaceutical properties, (e.g., pharmacokinetics (PK) and bioavailability) and safety. This led to a series of initiatives in pharmaceutical organizations to profile the properties of compounds earlier in the R&D process, in order to reduce the negative impact of poor properties on successful drug development. An early initiative was to extend traditional development studies on drug metabolism, pharmacokinetics, and pharmaceutics into the late discovery phase. This was successful in keeping candidates with poor properties from entering development. However, it was then recognized that efficiency in discovery could be improved if properties were monitored throughout discovery, so that years of work were not wasted on candidates that would fail in late discovery.
Since properties have been measured during discovery, organizations have realized the magnitude of the effect of properties on success. For example, properties play a major role in penetration of drugs into the brain through the blood-brain barrier (BBB). Also, low in vivo bioavailability can be predicted early and appropriate steps taken to correct the problem. Activity measurements using enzymes, receptors, or cell-based models can be more accurately planned, performed, and interpreted, and inaccuracies or inconsistencies can be reduced. Thus, properties contribute not only to development success, but properties also affect success in discovery. Ignoring properties during discovery can lead to higher costs, longer timelines, and lower quality drug products. Early evaluation can increase discovery and development efficiency.
Key decisions had to be made by each pharmaceutical company on how to implement property measurement in discovery. Should traditional in vivo PK studies be moved into discovery, or should fundamental properties that contribute to the complex PK process (e.g., solubility, stability) be measured? In making this decision, a review of the requirements of discovery can guide the selection of a strategy for property profiling. Drug discovery studies typically involve the study of tens of thousands of compounds per year. Even if the number of compounds selected for in vivo PK testing is 1% of this total, then hundreds of compounds must be studied, involving thousands of in vivo dosings and tens of thousands of time point analyses. Medicinal chemists in discovery would always prefer data on more compounds in order to obtain more definitive information to guide lead selection and optimization, and 1% may not be sufficient. In addition to the number of compounds, discovery also demands short turnaround times, on the order of a couple of weeks. Also, the amount of material available for each compound is typically in the tens of milligrams. PK studies would consume all of this material.
Because of these limitations of compound number, turnaround time, and amount of material, many pharmaceutical organizations have decided that in vitro testing of fundamental properties is a better fit for discovery. Automation techniques that were developed for activity screening (e.g., high throughput screening) are readily adaptable for in vitro pharmaceutical profiling assays. Multiple fundamental property studies can be performed in a rapid manner, in parallel, for tens of thousands of compounds using milligram quantities. Furthermore, information on fundamental properties is very useful to medicinal chemists in discovery, because they provide insights that directly apply to planning structural modifications that can improve properties. These structure-property relationships (SPR) parallel the traditional structure-activity relationships (SAR) and synthetic modifications in medicinal chemistry. The automation of pharmaceutical property assays for discovery is discussed in this article.
As a result of access to this data, medicinal chemists have been able build a balanced approach to candidate discovery, involving both activity and properties in a yin-yang relationship. Where SAR was previously the driving force in discovery, this balanced approach has resulted in compounds that occupy both acceptable activity and property space. Typically, the best candidates for development do not have the highest activity or best properties, but incorporate acceptable levels of each, to result in the best pharmaceutical agent for clinical use.
Selection of Properties to Profile during Discovery
There are many properties that can be assayed using higher throughput in vitro assays. While all of this data can be useful, resource limitations require a wise selection of which properties will have the greatest impact on drug discovery teams. Three concepts can contribute to this selection process: (1) examine the in vivo barriers that a compound faces in moving from the oral dosage form to the therapeutic target protein in the tissues, (2) examine the barriers that a compound faces with in vitro bioassays or in the laboratory, and (3) examine the properties that can be effectively calculated using software.
In vivo barriers 1 include stability (low pH in stomach and small intestine, enzymatic degradation in intestine and blood, metabolism in intestine and liver), solubility (aqueous pH from 2 to 8, gastric intestinal fluids), permeability (lipid membranes, BBB, efflux, active transport), protein binding (plasma), and distribution (tissues). In vitro and laboratory barriers include stability (aqueous and dimethyl sulfoxide [DMSO] solutions, photolytic degradation, oxidation, moisture, bioassay media), solubility (aqueous and DMSO solutions, bioassay media), permeability (cell membranes), and protein binding (bioassay medium). By targeting the barriers, property assays can be selected that provide insight on the performance of each compound at a barrier which is primarily governed by that property. The additional properties of pKa and lipophilicity are fundamental contributors to solubility, permeability, binding, and distribution. Advances in software modeling have resulted in higher quality predictions for properties like pKa and lipophilicity in recent years.
It is beyond the scope of this article to describe all of the assays that have been developed for property screening. In-depth reviews of physicochemical assays 2 and metabolic assays 3 are available. This article describes automation of key assays that have been selected in labs as a result of our experience and evaluation of cost and impact. In our labs, we have selected: LC/UV/MS integrity, parallel artificial membrane permeability assay (PAMPA), direct UV solubility, microsomal stability, and fluorogenic cytochrome P450 (CYP) inhibition. These provide examples of automation choices for in vitro high throughput pharmaceutical profiling that are commonly employed in drug discovery (Table 1).
Examples of automation in pharmaceutical profiling
Automation of Pharmaceutical Property Assays
Integrity
Integrity addresses the structure and purity of the sample that is being tested in discovery. Compound libraries used in discovery have been assembled from many sources, so it is good to assure that SAR is based on correct structures and that activity is produced by the putative compound and not an impurity. Compounds may have been synthesized decades ago and could have degraded during storage. Compounds may have degraded while they were dissolved and undergoing freezing and thawing cycles, or exposed to atmospheric water or oxygen. 4 Occasionally, compounds are mishandled and mislabeled, resulting in activity being ascribed to the wrong structure. Impurities may produce activity in bioassays or interfere with property measurement. Integrity and purity data should be generated before compounds undergo testing for activity or properties.
Compound integrity is usually assayed using an LC/UV/MS system. 5 In this assay, a compound is first dissolved in an appropriate solvent that solubilizes all of the sample and leaves no turbidity or precipitate. Samples are placed in a well plate (96 or 384 well) and sequentially injected using a high performance liquid chromatography (HPLC) autosampler. Sample components are separated by the HPLC column, which is typically operated over a wide mobile phase gradient to accommodate compounds with diverse polarities. Sample components are detected and their relative concentration is measured using a UV detector. An on-line mass spectrometer, typically with electrospray or atmospheric chemical ionization, produces spectra from which the molecular weight of the sample components can be obtained for confirmation of the identity of the sample.
Automation has been used so long with HPLCs that we sometimes take it for granted. Compared to manual syringe injection, autosamplers allow the reliable unattended injection of hundreds of samples from 96 or 384 well plates. The system software automatically controls all aspects of instrument operation, produces data outputs that can be rapidly reviewed by the analytical chemist, and provides a summary report that readily interfaces with Microsoft Excel or other data analysis packages. This instrumental and software automation reduces the human resources needed for the assay to less than 10% of the manual activities that would be otherwise required. LC/MS instruments 6 are widely used in drug discovery and represent powerful systems that are integrated via automation.
Permeability
A drug molecule must pass through numerous lipid membranes to move from the gastrointestinal (GI) track to the therapeutic target tissue. This is important to the absorption of orally administered drugs. In cell-based discovery bioassays, compounds must pass through the cell membrane to reach an intracellular target. There are several mechanisms by which molecules can penetrate lipid membranes or be excluded. The most important of these is passive diffusion, but paracellular, active uptake, and efflux mechanisms can be important, depending on the membrane and the compound.
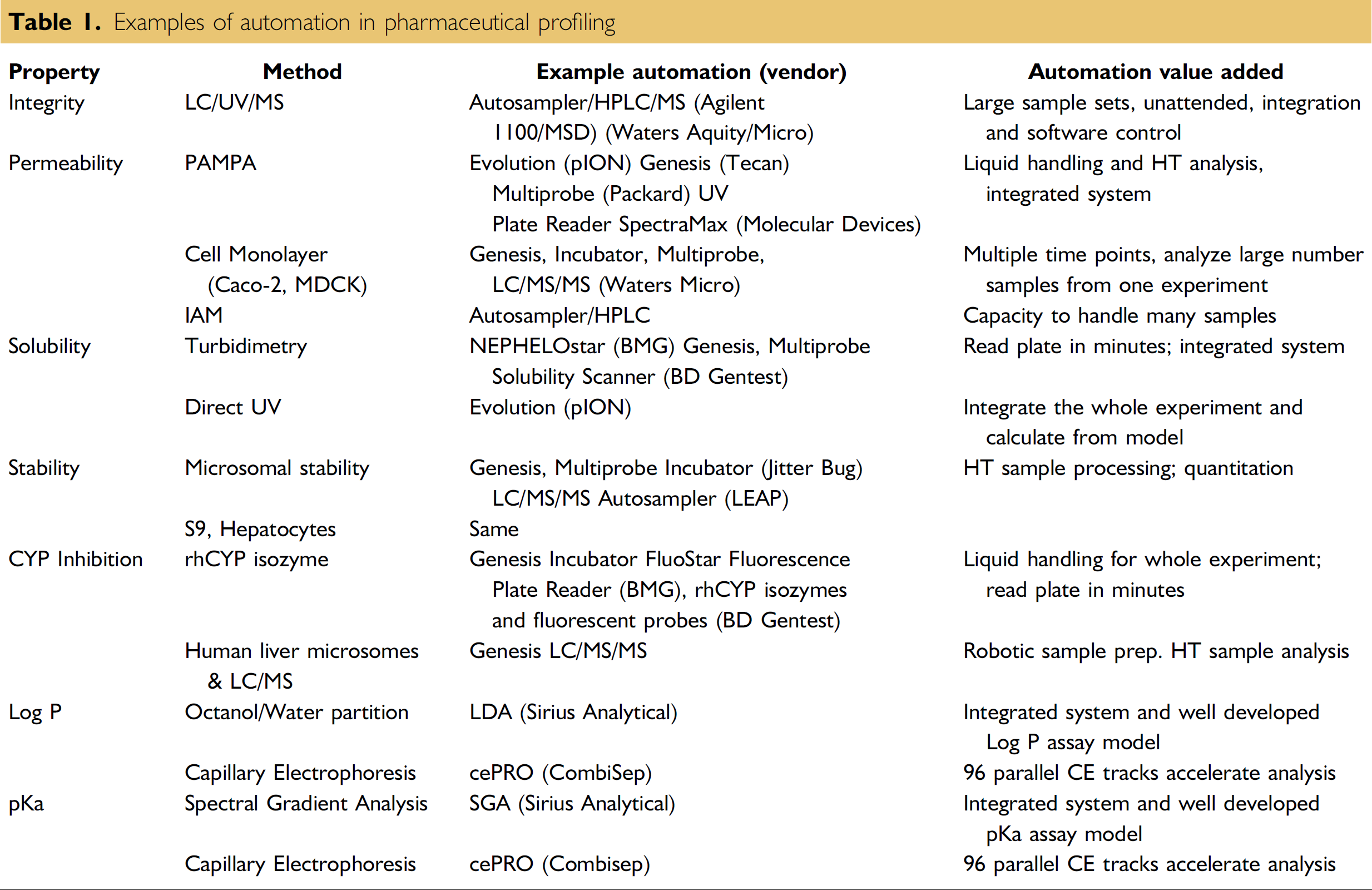
In vitro methods for permeability include: PAMPA, which measures only passive diffusion, cell monolayer methods (e.g., Caco-2, MDCK), and immobilized artificial membrane (IAM) HPLC chromatography.
IAM 7 utilizes a phospholipid covalently bonded to silica support, and permeability is calibrated using retention time (Fig. 1). The automation of IAM uses standard HPLC autosamplers and software. Generally, the correlation for PAMPA and cell monolayer methods to in vivo permeability is better than for IAM.
Examples of automated permeability assays.
PAMPA 8 was developed with automation in mind. The 96 well format is readily automated (Fig. 1). Samples are usually delivered to the robot as DMSO solutions and are diluted in buffer. A 96 well plate is filled with test compound in buffer at about 50 ug/mL and is the donor. On top of this is placed a 96 well filter plate, and the pores of the filter are filled with phospholipid diluted in organic solvent (e.g., dodecane). Blank buffer then fills the wells of the filter plate, to become the acceptor, and the permeation experiment begins. After the conclusion of the incubation, buffer is removed from the donor and acceptor wells, transferred to a UV plate and analyzed using a UV plate reader for quantitation of the concentration. Variations of the method involve (1) membrane lipid composition (e.g., brain lipid for BBB, 9 lipid for GI 10 ), (2) pH of the buffers, and (3) additives to the acceptor well to create a “sink” condition 11 and agitation of the donor buffer to reduce the unstirred water layer.
Automation of the PAMPA experiment is straightforward. Liquids are readily handled using multichannel pipetters on a robotic platform (e.g., Tecan Genesis, Packard Multiprobe). A commercial product (Evolution) has been developed by pION (Woburn, MA), including an in-depth understanding of the PAMPA experiment and model for accurate calculation of the PAMPA data. The sophisticated understanding of a method like PAMPA and incorporation of this model into the software, which controls the instrument and calculates the results, is a point that is often overlooked by scientists who attempt to develop their own instrumentation. pION and Millipore Inc. have developed specialized donor and acceptor plates that fit well together and have minimal nonspecific binding that can compromise the assay.
Cell-monlayer permeability 12,13 automation is similar to PAMPA. Both donor and acceptor solutions are sampled. Millipore and Corning Costar have developed specialized “transwell” plates that facilitate cell culture and allow the well below the cells (basolateral) to be sampled robotically without removing the upper wells (apical). Because of the time required for cell culture maintenance (21 days cell culture for Caco-2, 3 days for MDCK), a robotic system has been developed for automated cell maintenance and incubation (Tecan).
Solubility
Solubility is also a crucial factor for in vivo bioavailability. The compound must dissolve and diffuse to the GI epithelial cells in order to be absorbed. Solubility is also important for in vitro bioassays, and lack of full solubility at the test concentration will cause underestimation of the compound's true activity.
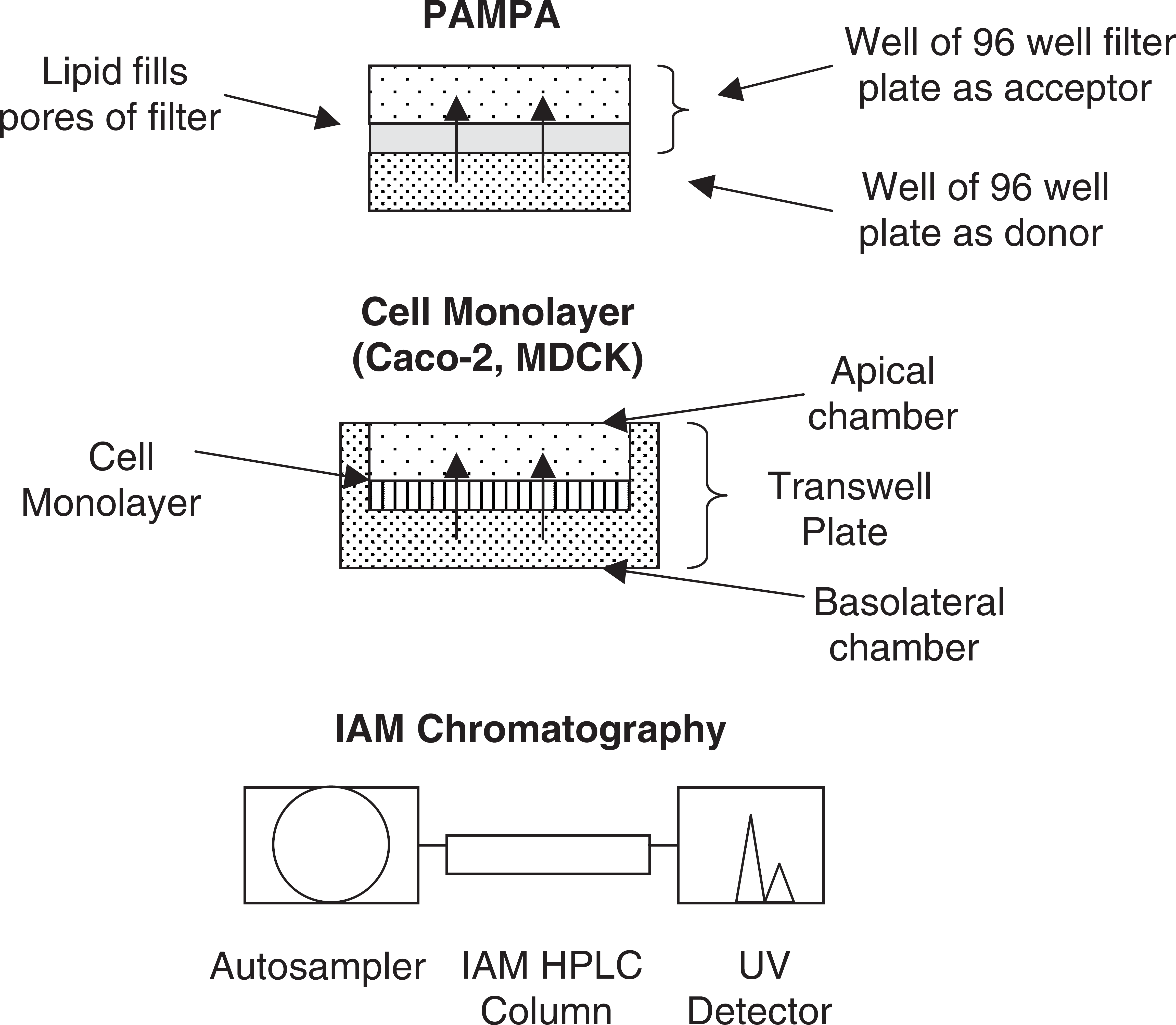
There are two types of solubility experiments that are performed (Fig. 2). In the first experiment, the drug material is first dissolved in an organic solvent, typically DMSO, at about 10-20 mg/mL, and then a small volume is added to an aqueous buffer. After a period of time, undiluted material is removed and the concentration of compound is measured as an estimate of the maximum solubility of the compound under these conditions. This experiment is termed “kinetic” solubility. In the second experiment, aqueous buffer is added to solid drug material and then incubated and measured as above. This experiment is termed “thermodynamic” or “equilibrium” solubility. In the thermodynamic experiment, the compound must dissolve by overcoming any crystal forces in the solid state, and in the kinetic experiment these have been overcome prior to the experiment by DMSO dissolution.
Examples of automated solubility assays.
The kinetic experiment appears to be best suited to discovery, where nearly all experiments are conducted by first dissolving a compound in DMSO. Furthermore, compounds are formulated with solubilizers for discovery in vivo dosing experiments. In addition, in medicinal chemistry, labs solids are produced by evaporating solvent. This creates a complex amorphous solid of unspecified crystal and noncrystalline forms. Because the transition from solid state to dissolved state is crucial for the thermodynamic experiment, and the method of solid preparation varies from lab to lab, thermodynamic solubility appears to be an unreliable approach for rank ordering compounds in discovery. Compounds of continuing interest will have subsequent batches prepared and salt forms and formulations will be investigated, all of which will have different thermodynamic solubility. While thermodynamic solubility is useful when preparing a data package for a proposed development candidate, kinetic solubility seems most appropriate for high throughput in vitro assays in discovery. There are two common approaches for automating kinetic solubility assays: the “direct” method and the “turbidimetric” method.
In the direct method, 14 a small volume of concentrated DMSO solution is added to a well containing buffer. The solution is allowed to equilibrate for 1 to 24 h. Then the solution is filtered using a filter plate and manifold, which are commonly used for pharmacokinetics analysis of plasma samples following protein precipitation. The solution can also be centrifuged at high speed to pellet the undissolved material. The supernatant is assayed using a UV plate reader or HPLC. pION has developed an integrated system for solubility (Evolution) that uses a Tecan Genesis robot equipped with a manifold for filtering, a Molecular Devices UV plate reader, and customized software that operates the system and calculates solubility using a mathematical model. They have also patented a method for using cosolvent to dissolve low solubility compounds for a reference standard.
In the turbidimetric method, 15 a small volume of concentrated DMSO solution is added to a well containing buffer. This solution is serially diluted across other wells and the plate is allowed to equilibrate. The plate is taken to a plate reader that measures light scattering and the series of wells are scanned. The wells in which the compound is not soluble are turbid and produce light scattering, whereas the wells in which the compound is completely dissolved have no turbidity. A plot of turbidity vs. concentration indicates the maximum concentration dissolved, which is taken as the solubility. BMG Labtechnologies has developed a widely used turbidimetric solubility method which features their NEPHEL Oostar instrument with laser light scattering on a well plate. BD Gentest has developed a Solubility Scanner instrument that, instead of laser light scattering, uses technology from flow cytometry to detect turbidity.
Both of these methods are very amenable to lab automation involving liquid handling and well plate manipulations. Solution conditions are very significant in the experiment. pH is important, because compounds exhibit a solubility-pH profile, which is a function of the intrinsic solubility of the neutral form, the solubility of the ionized form (protonated for bases and deprotonated for acids), which is typically much higher, and the pKa(s) of the compound. DMSO concentration is also important, because DMSO can operate as a cosolvent, which changes the dielectric constant of the solution and helps to solvate the more lipophilic compounds.
Stability
There are many aspects of compound stability that play a role in discovery success. Most notably, metabolic stability is a crucial factor for in vivo bioavailability, because many compounds are rapidly cleared in vivo by first pass metabolism. Compounds in the GI are subject to hydrolysis and enzymatic oxidation in the epithelial cells, and once they are absorbed, are immediately transported to the liver via the portal vein where they are subjected to various metabolic enzymes. Of greatest interest is cytochrome P450 (CYP) oxidation. Compounds (e.g., esters, amides, carbamides) are also subject to hydrolytic enzymatic degradation in the GI and blood stream, which is often estimated with an assay that uses plasma incubation. 16 Other stability issues include stability in aqueous solutions of various pHs and composition, 17 and exposure to water, oxygen, and light in the solid or solution states.
Oxidative metabolic stability is widely assayed using liver microsomes, which are prepared from tissue by homogenation and centrifugation. Several vendors supply microsomes from various species that can be used like reagents. The user should be careful to verify the activity of each new batch. More detailed studies make use of individual isozymes of CYP, which are produced recombinantly and used to determine the actual isozymes that are responsible for metabolism of a compound. HT microsomal stability assays typically use a single generic species (e.g., rat) and then perform customized studies using selected species that are of greater relevance to the particular project. Because it is an enzyme assay, cofactors and buffer specifications should be carefully followed in assays. Nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) is needed as an electron source. Some users just add NADPH when the incubation is short, but others use NADPH regenerating mixture that produces NADPH from an enzymatic reaction.
There are many conditions that should be carefully controlled in the assay. These include substrate concentration, DMSO concentration, microsome protein concentration, and microsome preparation. Many users have adapted literature methods to their own laboratories; however, there is a major effect of conditions on the data produced by the assay. Conditions and robotics of microsomal stability have been studied to provide an optimal method that is reproducible. 18,19 It is also important that users run quality control standards along with each assay plate to insure that the assay is performing properly. Quality control standards of several levels of stability are recommended.
As with other automated pharmaceutical profiling methods, the samples are brought to the robot in a stock plate dissolved in DMSO. One useful procedural element is to dilute the microsomes in buffer prior to use, so that they are less viscous for the pipetters to accurately measure. 18 We have elected to produce a time equals zero sample and a time equals 15 min sample in duplicate for each compound, based on a mathematical analysis of the method. 19 The reaction is stopped by adding cold acetonitrile and the majority of the protein is removed by centrifugation. The supernatant from the incubated samples is analyzed using LC/MS/MS with a rapid generic gradient mobile phase program over 3 min. The initial portion of the HPLC eluant is diverted to waste to reduce contamination of the MS ion source. Because about 45 different compounds are being analyzed per plate, the development of sensitive MS/MS conditions for analysis would be very time consuming if they were performed manually. Mass spectrometry companies have developed automated procedures (e.g., Waters QuanOptimize) that develop very good conditions by optimizing ionization polarity (+ or −), ion source cone voltage, parent-daughter ion pairs, and collision energy for MS/MS analysis. Automated MS/MS condition selection can be done at the same time as sample incubation, and preparation is being done on the robotic platform (e.g., Packard Multiprobe) so that the instrumental analysis is ready when the samples are ready. We have also found that the use of high capacity, high speed, and rugged autosamplers, such as the LEAP CTC Pal with Cycle Composer software, is a very dependable way to analyze samples from hundreds of compounds per day. It is also possible to eliminate the HPLC column for faster analysis (Janiszewski, Kerns). 20,21 Samples are injected onto a reverse phase trap and back flushed directly into the MS/MS. Trapping eliminates the proteins and salts from interfering with the electrospray interface, and the analysis can be performed in 1.2 min versus 3-4 min that are required with an HPLC separation. Selectivity is sufficient using MS/MS.
Variations on the incubation conditions can be used to investigate specific metabolic properties. For example, another liver preparation, S9, which contains more metabolizing enzymes than microsomes can be used, or uridine diphosphoglucoronic acid can be added to produce glucuronide conjugate metabolites to examine Phase II metabolism. Glutathione may be added to a microsomal assay to investigate the reactivity of the metabolites, which might cause toxic reactions.
Conditions for solution stability have also been developed. 17 This method would be useful for assaying the stability of compounds in pH buffers, assay buffers, cell culture buffers, and simulated gastric intestinal fluids. Advantage can be taken of the ability of some HPLC autosampler syringes to add solutions of reagents to samples in well plates in the autosampler and to incubate the samples at a specified temperature (e.g., 37 °C). The autosampler is programmed to inject aliquots of this reaction mixture into the HPLC at specified time points, in order to calculate the reaction kinetics. Again, the automation of the autosampler system and software allows unattended experiments over many hours or days for large numbers of samples.
CYP Inhibition
Several pharmaceutical products have been withdrawn from the market because of their tendency to cause drug-drug interactions (DDI) with another coadministered drug. The cause of DDI is the competition of both drugs for the same enzyme or transporter. One major area of competition is for specific isoenzymes of CYP in the liver. If one drug inhibits metabolism of a second drug, then the second drug may build up to a toxic level. For this reason, screening is often performed during discovery to determine if a compound is a potent inhibitor of a key CYP isozyme.
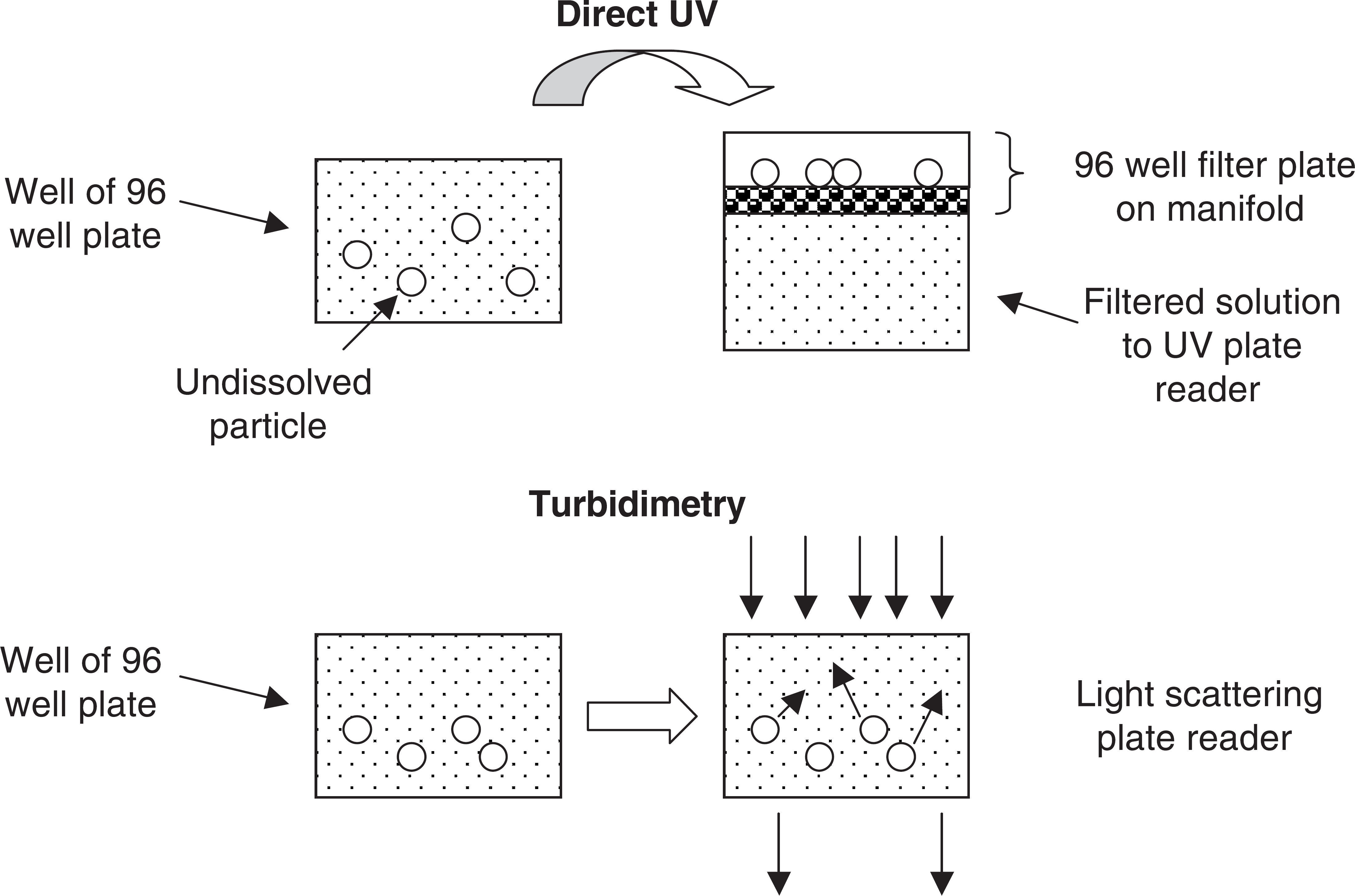
High throughput CYP inhibition assays (Fig. 3) are often performed using a “probe” substrate that becomes fluorescent when metabolized. Co-incubation of the probe and the test compound indicates whether the test compound inhibits the production of fluorescence from the probe compound. Like the metabolic stability method, this assay is readily automated on laboratory robotics platforms. The assay uses the same reagents as the microsomal stability assay, except that the CYPs are recombinant human CYP isozymes. The rhCYP isozymes and special probe compounds can be obtained commercially (e.g., BD Gentest). Fluorescence is measured using a standard 96 or 384 well plate reader (e.g., BMG FluoStar).
Examples of automated CYP inhibition assays.
Another CYP inhibition assay method uses actual drug compounds as probes, and their metabolism to hydroxylated forms is monitored using LC/MS/MS. Often, human liver microsomes are used, which contain all of the CYP isozymes in their natural abundance. Incubations are performed in a similar manner to the fluorometric method on a robotic platform.
Lipophilicity and pKa
Permeability and solubility measurements parallel in vivo properties that have a great impact on absorption of orally administered drugs. However, the more fundamental physicochemical properties lipophilicity and pKa contribute to permeability and solubility and are often measured during drug discovery.
Lipophilicity affects the distribution of a molecule between a lipid or nonpolar environment and an aqueous or polar environment. It is often measured as the equilibrium distribution of a compound between water and octanol and is termed Log Pow. Compounds with higher Log P tend to have lower aqueous solubility and higher membrane permeability. Automation of Log P measurement can be a difficult task. Analiza Inc. developed the AlogPW workstation that integrates the entire octanol-water partition assay in one system. Another approach for lipophilicity is reversed phase HPLC, which simulates the partitioning behavior in a convenient automated system. 22,23 Sirius Analytical developed the ProfilerLDA workstation that integrates the entire Log P assay in one system, using a variation of HPLC with octanol saturation. One high throughput strategy for using HPLC for lipophilicity involves the cochromatography of compound mixtures. 24 The capacity of the HPLC format is not saturated. The total analysis time for a set of compounds by “combinatorial analysis” is reduced to a fraction of the time of sequential analysis.
The ionization of molecules in aqueous solution is affected by pKa; it is measured by the change in ionization with pH. A basic compound with a pKa of 9 will have an equal distribution of protonated and neutral molecules at pH 9, with protonation increasing at lower pH and neutral species increasing at higher pH. An acidic compound with a pKa of 4 will have an equal distribution of deprotonated and neutral molecules at pH 4, with neutral species increasing at lower pH and deprotonated species at higher pH. Sirius Analytical has developed the ProfilerSGA workstation that integrates the pKa assay in one system, using the change in UV absorption of a molecule as it changes in ionization state.
With the availability of highly developed software for lipophilicity and pKa, some organizations have decided to calculate these properties, thus saving human and automation resources for other assays. In selecting an in silico approach, a company must carefully consider the cost of the software, the cost of the automation, FTEs, quality of the results that are needed, and validation of the in silico or in vitro tool using in-house compounds.
Other Key Aspects of Assays That are Consistent with Automation
When the automated methods have been implemented, there are automation steps that can be added to the organization' infrastructure to make the operation run efficiently. These can be determined by examining the laboratory' workflow: analyses are requested, samples and plates are prepared and tracked, assays are performed, data is reported, and conclusions are presented.
Analysis request and sample tracking can require a lot of time and effort. It is useful to have a computerized tracking system that assists individual analytical chemists with planning and completing their work. A tracking system that is maintained on a central network computer helps everyone manage their work and assure that they are delivering data in a timely manner. Metrics from such a system can help the group evaluate their performance and improve their workflow to aid the speed of the organization. Typically, such tracking systems are constructed in-house according to the needs and responsibilities of the group.
Pharmaceutical profiling produces large amounts of data. For thousands of compounds per year there may be 5-10 separate measurements. Moreover, these data have the greatest impact for the organization if they are made widely available to scientists in the company. However, preparation and distribution of reports can be very time consuming. Most companies have found that the best ways to deal with this is by using a central database. It is very effective to upload data from a table (e.g., spreadsheet) using a program that places the data in the proper fields in the central corporate database (e.g., Oracle tables) via the in-house network. Interface programs are relatively simple for an experienced programmer to construct. The automation of this upload procedure, compared to manual data entry, saves considerable time and effort and is very reliable compared to transcription errors that can occur.
Inevitably, the scientists that generate the pharmaceutical property data need to become experts in the properties and application of the data for discovery. This is because experts of this type have existed in development, but not in discovery. Furthermore, they are called upon by project teams to discuss their data and apply them to the project at team meetings or at individual conferences with key project leaders. An effective approach for presenting the data is very useful in effective communications. One approach is to tabulate the data and color code it according to ranges of favorability. For example, in microsomal stability a t1/2 of less than 14 min may be considered low and colored red, a t1/2 between 14 and 60 min may be considered moderate and colored yellow, and greater than 60 min may be considered high and colored green. Programs may be prepared in Visual Basic or another language to convert spreadsheets to attractive report tables with color coding of cells according to the data values in the cell. Tables allow project leaders to quickly review the properties of the latest compounds and to rapidly identify, via color coding, the compounds with favorable and unfavorable properties, and their trends. Presentation of data at meetings can be very effective if it is related to structures. Medicinal chemists use structure-activity relationships very effectively in driving the optimization of activity by making structural modifications. In the same manner, presentation of structure-property relationships allows chemists to visualize structural modifications to optimize properties.
Application of Pharmaceutical Profiling Data in Drug Discovery
Property data can have an important impact on drug discovery in several ways. Because properties affect discovery bioassay and property assays, as well as development success, we should insure that the data is used for discovery experiments. For example, special care should be exerted in preparing aqueous stock solutions of low solubility compounds, and biochemists should watch for precipitation of compounds in assays. The IC50 curves that are generated should be reviewed for irregular shapes that indicate that solubility has limited accurate measurement of the activity.
It should also be recognized that activity and property measurement are similar and parallel activities. Each uses in vitro assays for higher throughput assessment of large numbers of compounds with various assay formats. Each also selects the best compounds for in vivo dosing to determine either in vivo efficacy or in vivo pharmacokinetics and exposure. This parallel format should allow parallel data generation for parallel optimization of activity and properties. Furthermore, the results of these assays allow redesign of the structure to improve performance, synthesis of the new series analog, and testing of the new compound using activity and property assays in an iterative cycle leading to lead optimization.
Future Directions
Pharmaceutical profiling is still relatively young in the lifecycle of discovery technologies. It needs to continue improvement in quickness, quantity, and quality.
Higher levels of automation are also being implemented. The progression of technologies has been from manual liquid handling (pipetters) to lab robotics (multiprobe robots). The next step is to integrate several robotic platforms around a central articulated arm robot. 25,26 This system performs the manual movements that a scientist would normally have to do, its software tracks the several processes operating around the central track, and the robotic arm arrives just in time to perform the next scheduled task. Such systems are expensive and typically require considerable time to set up, but in the long term they reduce the human resources necessary to perform the assays and increase throughput.
As high quality data are generated, the software models continue to improve. When the predictions become reliable, it is cost effective to retire the assay and move resources to another assay that is needed. It is important that the software be internally validated to insure that reliable values are being generated on which decisions can be based.
Conclusions
Pharmaceutical profiling has greatly benefited drug discovery research by providing insights on improving the delivery of compounds in higher quantity to the therapeutic target in the tissues, and reducing toxicity. The data have also benefited the planning and interpretation of discovery activity experiments. There are many ways that automation contributes to pharmaceutical profiling (Table 2): robotic liquid handling, high capacity detection, and data processing and reporting (Figure 4). Automation has greatly contributed to this effort by raising the productivity per FTE and improving the distribution of this data throughout the drug discovery group. These trends will continue as pharmaceutical profiling becomes further ingrained as an integral function in drug discovery.
Summary of automation in pharmaceutical profiling

Crucial elements of automation for pharmaceutical profiling.