
Abstract
A single nucleotide polymorphism (SNP) scoring assay that uses ligation-dependent Rolling Circle Amplification (RCA)† was transferred to a series of automated protocols addressing a range of throughput levels. The systems utilised various automation modules consisting of custom-made and off-the-shelf devices. Several system parameters were evaluated to ensure assay integrity and homogeneity. These included reagent carry over, liquid evaporation rates, thermal regulation of reactions and fluorescence reading capabilities.
Data analysis software was developed in order to rapidly allocate SNP calls from data generated by the automated system. A modified fuzzy c-means clustering algorithm was employed to separate data points into groups associated with specific genotypes. Data were then presented graphically and within a summary table, which allowed easy and rapid organization and interpretation of data.
Keywords
Introduction
An increasing number of known sequence variants now promise to shed light onto the genetic determinants of biological processes. The assignment of these different variants in an otherwise conserved DNA region is used for the identification of markers associated with human diseases or pharmacological responses. The majority of this sequence variation consists of differences at single nucleotide sites referred to as single nucleotide polymorphisms (SNPs). The vast majority of SNPs are bi-allelic with the least frequent allele having an abundance of 1% or greater. 1 Due to their abundance and stability, SNPs are anticipated to represent a powerful tool in pharmacogenetics and diagnostic applications. A full genome linkage scan can be achieved with 900 SNPs, however 2000–3000 SNPs would provide more detailed information. 2 In association studies, approximately 100,000 SNPs would have to be screened in order to investigate subtle genetic influences in complex disorders. 3 Due to the vast numbers of genotypes that are required for population analyses, automation and organization of SNP determination methodologies is essential. Applications of SNP analyses thus extend from investigations of a small number of SNPs known to be associated with specific diseases to whole genome scans of large populations. To address these requirements, technologies aimed at providing flexible and scalable solutions to SNP determination have been investigated. This paper describes several systems developed to meet various throughput requirements for SNP analysis using a ligation dependent rolling circle amplification (RCA) SNP scoring assay. 4 –6
The RCA method exploits open circle probes (OCPs), linear oligonucleotides comprising an allele specific coded backbone and SNP-specific arms (Figure 1a). DNA fragments spanning the SNP site, generated by PCR amplification of genomic DNA, is heat denatured to form single strands. The OCPs and denatured PCR fragments are incubated together in order, so that the juxtaposed arms of the OCP hybridize to the allele. In the presence of a thermostable ligase, the OCP forms a circular template (Figure 1b) at 65°C, which can then be amplified. Amplification of a circularised probe by RCA requires two primers. The first primer hybridises to its complementary region on the probe backbone. At 65°C, thermostable strand-displacing DNA polymerases extend the primer, which eventually displaces itself at its 5' end, after one complete revolution of the circularised probe is made. Continued polymerisation and displacement result in the generation of a single-stranded, concatameric DNA copy of the original probe. The second primer binds to each tandem repeat of the first strand product. As these multiple priming events elongate, they initiate strand displacement, creating single-stranded DNA products which contain further binding sites for the first amplification primer; this is known as branching.
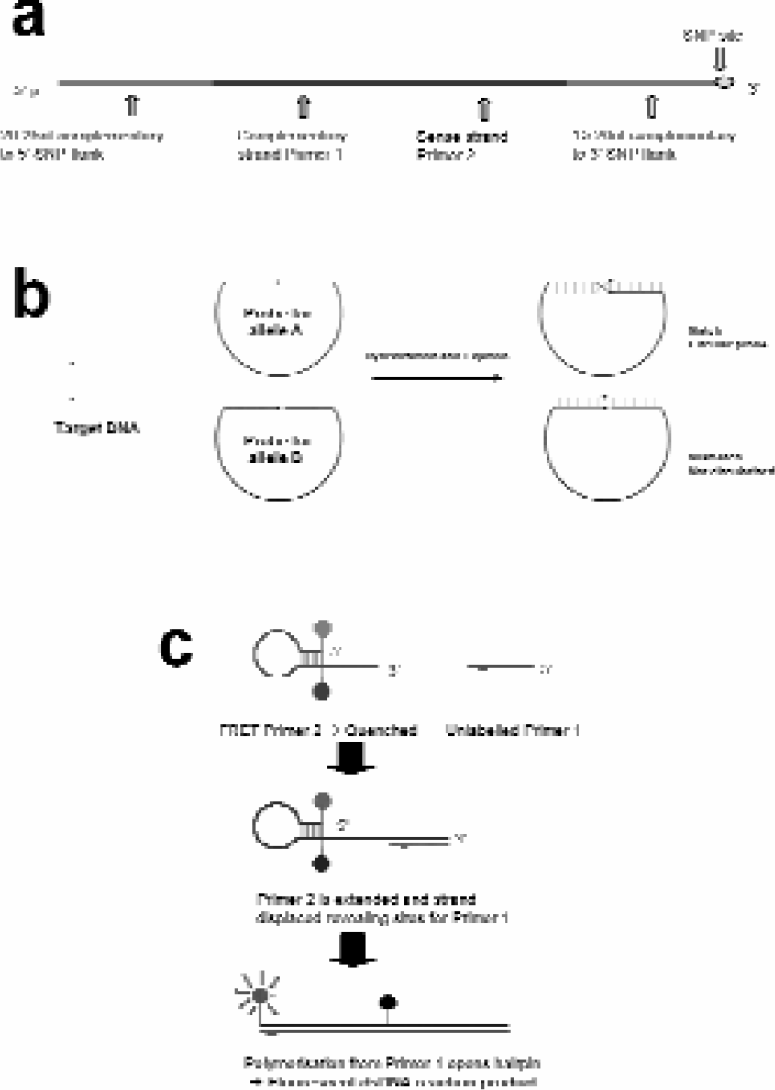
(a) Schematic representation of open circle probes used to interrogate DNA for SNP analysis. Each open circle probe consists of an oligonucleotide, 80–90 bases in length. (b) Mechanism of SNP specific hybridization and ligation resulting in circularisation of probes. (c) Endpoint analysis using Amplifluor probes.
Amplifluor™ technology 7 (Intergen, Purchase, NY, USA) is utilized for end-point detection of the amplified probe, enabling a homogenous assay format. 4 Amplifluor detection primers have a 5' hairpin loop, labeled with a fluorophore and a quencher. In its native state, the fluor and quencher are in sufficient proximity for quenching of the fluorophore to occur. By labeling the second RCA amplification primer with these hairpin structures, they become incorporated into the double-stranded RCA products. As the complementary strands to the hairpin primers are synthesised during branch formation, the polymerase displaces the stem and opens out the 5' end. In this extended conformation the fluor is no longer quenched and fluorescence can be detected (Figure 1c). The use of specific primers to each of the backbones labeled with two different fluors enables discrete detection of both alleles of a SNP in one reaction. The detection of two alleles in a single reaction is vital for accurate SNP calling because it eliminates miscalls of heterozygotes, since pipetting differences result in a failed reaction rather than a homozygous miscall.
The fluorescent endpoint, together with the isothermal nature of the homogenous assay, makes the assay amenable to automation.
Manual Format
The manual assay format was initially employed to optimise assay conditions and to provide control reactions for use with subsequent automated assays. Reactions were performed in polypropylene tubes using handheld pipettes for all liquid handling steps and thermocyclers for incubations. Upon completion of the reaction, the entire volume was transferred to 384-well, low-volume black polystyrene microtitre plates (MTPs) for fluorescence reading in a Tecan Ultra plate reader. Although this manual format provides a great deal of flexibility, the labour intensive liquid transfer steps limited the throughput to circa 96 reactions per day.
Medium Throughput
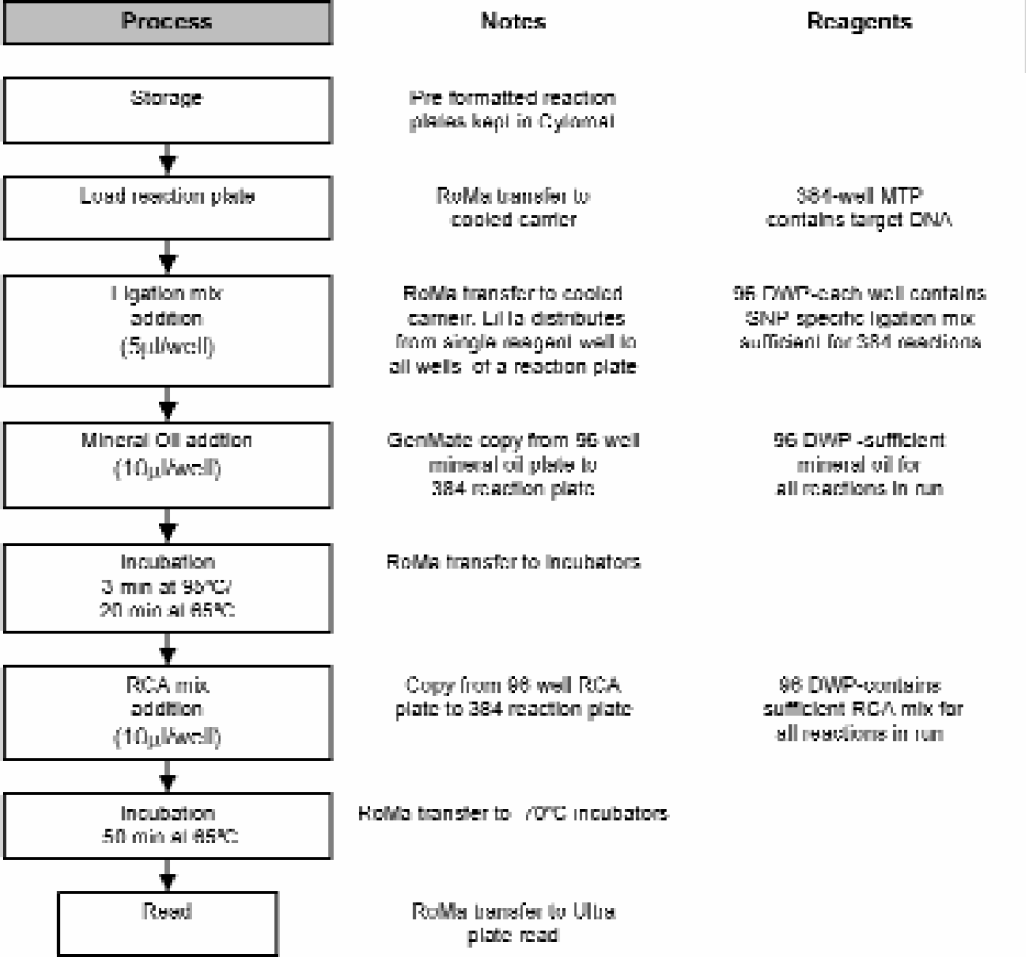
In order to achieve higher throughput, automated liquid handling steps for 384-well MTPs were developed. Since incubations were accomplished on thermocyclers, thin walled polypropylene polymerase chain reaction (PCR) MTPs were utilized. Black ABgene Thermo-Fast® mark II MTPs were employed. The use of these plates brought about several technical challenges to the various automated elements of the process. Figure 2 shows the process workflow with the various reagents and components employed.
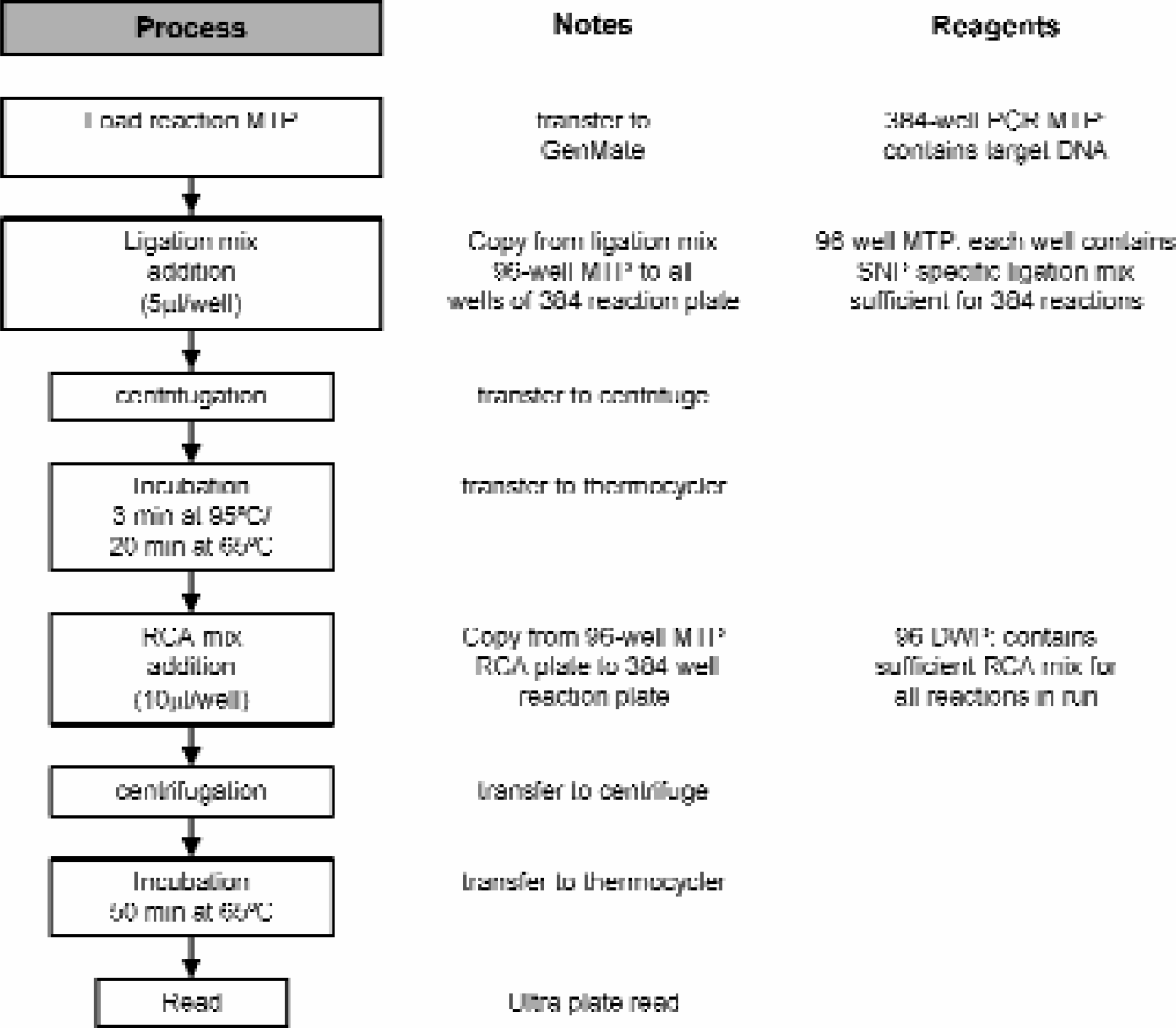
Process workflow and associated notes for medium throughput protocol.
To reduce possible cross contamination risks the target PCR products were generated in a separate laboratory. This approach is adopted by many automation laboratories, which often use dedicated systems for their PCR requirements. PCR products were prepared in 384-well MTP format. Each PCR MTP contained DNA from 384 different individuals amplified for the PCR product encompassing the same SNP site. A 96-tip fixed head pipetting station was used to aliquot 5μl of the PCR product into a Thermo-Fast reaction plate. A series of such 384-well MTPs containing the target DNA was prepared in the PCR laboratory and was the starting point for all automated protocols.
A stand-alone Tecan GenMate robotic module was used for the liquid handling processes. The 96-tip standard volume head, employing 50μl disposable tips (DITIs), was used to rapidly dispense ligation and RCA reagents into the 384-well reaction plate containing the target DNA. 5μl of ligation mix (containing ligase, ligation buffer and OCPs) was aspirated from a standard 96-well polypropylene MTP, then dispensed into the first quadrant of the reaction plate. Ligation mix was dispensed into the bottom of the well and hence directly into the target DNA solution. No mixing of reagents was done at this stage as it was assumed that the initial 95°C incubation would mix target and ligation mix sufficiently. Prior to addition of ligation mix to the next reaction plate quadrant, DITIs were washed using the GenMate active wash station. Each well in the ligation mix source plate contained the same OCP due to the specific formatting of target DNA in the reaction plate.
Due to the narrow well diameter of the PCR plates, it was found to be necessary to centrifuge plates before positioning them on the thermocycler, to prevent the formation of air cavities beneath the liquid surface.
RCA mix was added using the same DITIs. A single source RCA mix 96-well MTP was required since the mix is generic for all SNP reactions. 10μl was aspirated from the RCA mix plate, and then dispensed into the bottom of the reaction plate's wells. Five 10μl mixes (aspirate then dispense) were then immediately performed. Prior to addition of RCA mix into the next quadrant, the DITIs were washed in the active wash station.
Amplification levels by rolling circle have been measured to be 109–1012 fold. 5 This compares to 106–109 fold amplification by PCR. Therefore a small amount of well-to-well cross contamination can be amplified giving rise to aberrant fluorescent products. Despite the rapid geometric amplification that occurs during RCA, water washes employing the GenMate active wash station using 20 times the dispensed volume were sufficient to prevent cross-contamination products being generated (Figure 3). Serial dispensing of reagents into microtitre plate well quadrants containing reactions with different genotypes of the same SNP using the same disposable tips throughout the entire assay demonstrated no carry over of reagents, resulting in a 100% calling accuracy. Moreover, the last dispensed quadrant wells, which did not contain PCR targets, did not generate fluorescent products indicating that there was insufficient circularised probe carried over to initiate an RCA reaction that was initially devoid of a template. The same DITIs were subsequently used throughout the whole process on multiple reaction plates with no detrimental effects (Figure 9b).
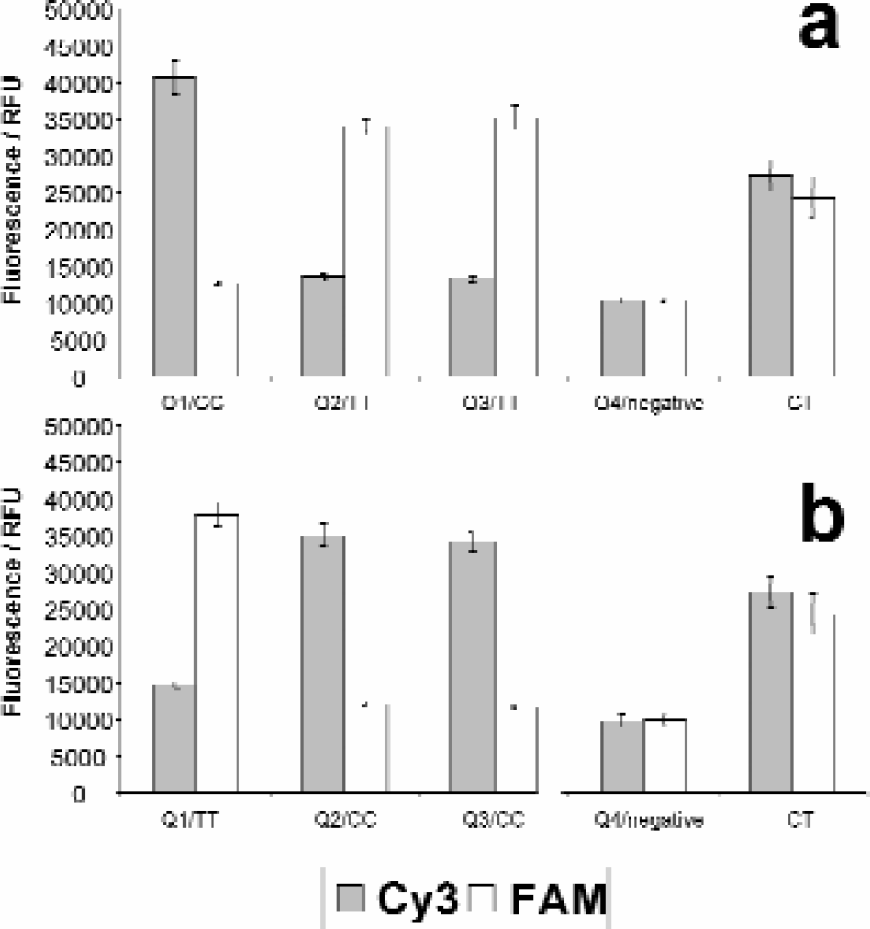
GenMate DITI cross contamination study. The same DITIs were used to dispense into the same quadrant wells of a 384 well microtitre plate in series (Q1, Q2, Q3 then Q4) to assess liquid carry over. Half the plate contained quadrants Q1, Q2 Q3 and Q4 with PCR targets genotypes CC, TT, TT and water only, respectively (a). In the other half of the plate quadrants Q1, Q2 Q3 and Q4 contained genotypes TT, CC, CC and water only, respectively (b). The plate was spiked with heterozygous targets in two of the negative wells. Error bars show standard deviation.
Liquid volumes undergoing incubations were between 10 and 20μl, thus evaporation control strategies were required for optimal reaction conditions. Unlike PCR, there is a second addition of reagents after an initial two-phase incubation. Whereas heat-sealing systems provide excellent seal integrity for standard PCR, resealing for a second incubation step can prove problematic. Furthermore, removal of seals for plate reading can be difficult unless foil strippers are used. As an alternative, adhesive foils were assessed and were found to decrease evaporation rates significantly, but the seals were found to be insufficient to completely prevent evaporation (Figure 4). Moreover, visual inspection of water filled wells indicated that well to well liquid migration occurred, often resulting in wells accumulating large amounts of liquid. This was due to the integrity of the seals being broken by plate expansion during heating — a phenomenon that is particularly notable with 384-well plates. However, effective sealing was observed with MJ Research ‘A’ seals, and visual inspection revealed that the intra-plate liquid migration was no longer apparent. The ‘A’ seals were therefore placed onto the reaction MTPs during each thermocycler incubation.
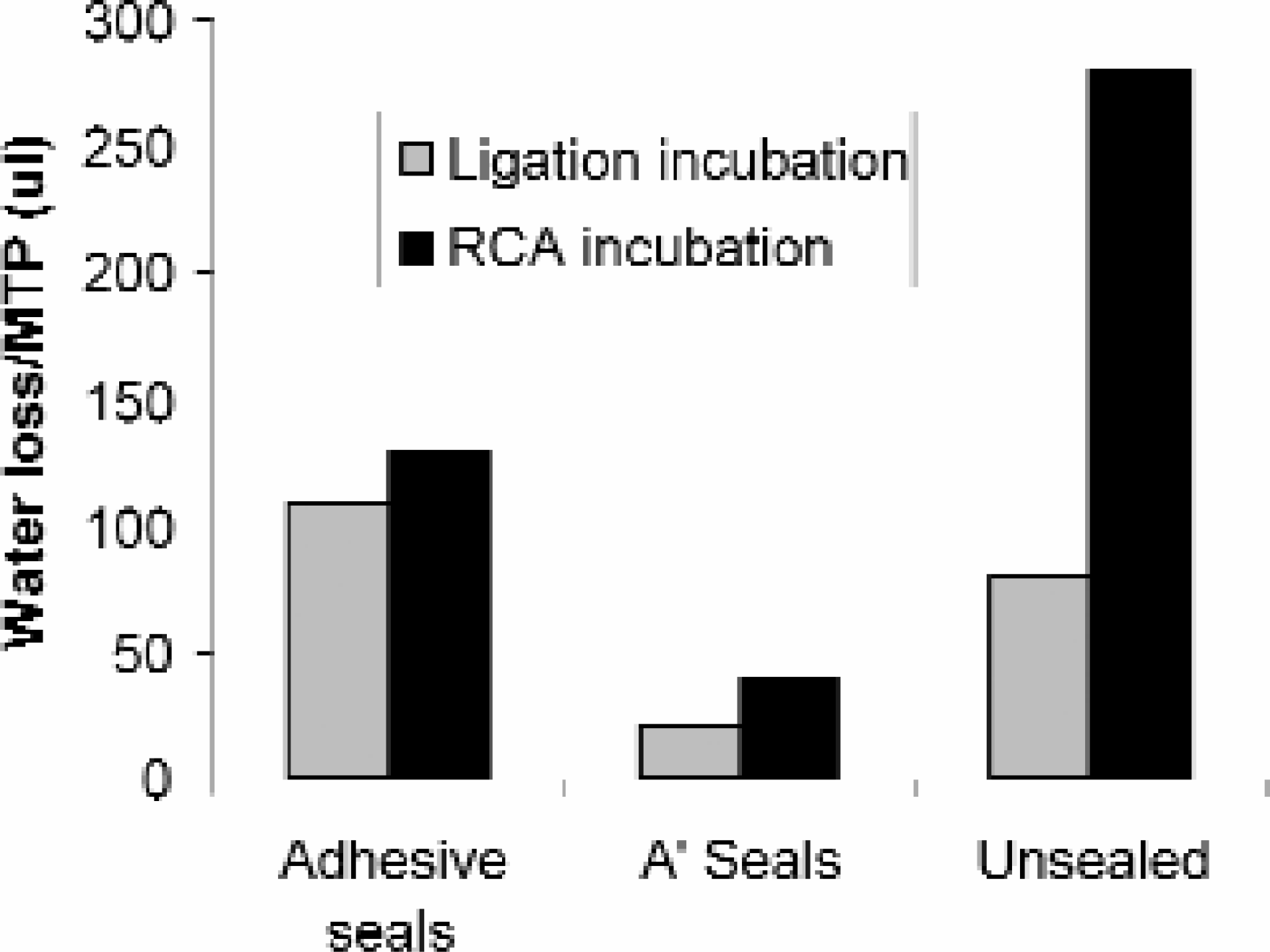
Liquid losses using adhesive and ‘A’ seals from 384 well PCR microtitre plates containing water incubated on a thermocycler. The two incubation periods simulate the conditions used in each step of the reaction.
Since the ‘A’ seals do not require a large amount of downward pressure from the heated thermocycler bonnet, the polypropylene plates were not severely distorted. The Tecan Ultra plate reader was consequently able to measure accurately fluorescence directly from the black PCR MTP without the need for transfer of reaction liquid into a low volume MTP. An ABgene robotic plate positioner was required to prevent bowing of the plate during loading into the Ultra.
Calculations based on real-time measurements of all processes including manual handling of plates indicate that 4600 SNPs (twelve 384-well MTPs) can be assayed by an individual worker in seven hours using four alpha blocks of a MJ Research Tetrad. A second Tetrad can increase the throughput to 6100 reactions (sixteen 384-well MTPs) in a similar time frame, transferring the process bottleneck from the Tetrad to the GenMate.
High Throughput
To increase throughput to higher levels, all processes were fully automated without the need for manual intervention. For this purpose an integrated multi-module approach was employed. A series of peripheral devices were interconnected around a central Tecan Genesis RSP200 liquid handling station (figure 5). The hardware consisted of a combination of off-the-shelf and custom components. These devices included:
Tecan GenMate
Tecan Ultra plate reader
Tecan customised 70°C incubators
Tecan customised 95°C incubators
Tecan customised cooling racks
Kendro Cytomat 4000 4°C cool store
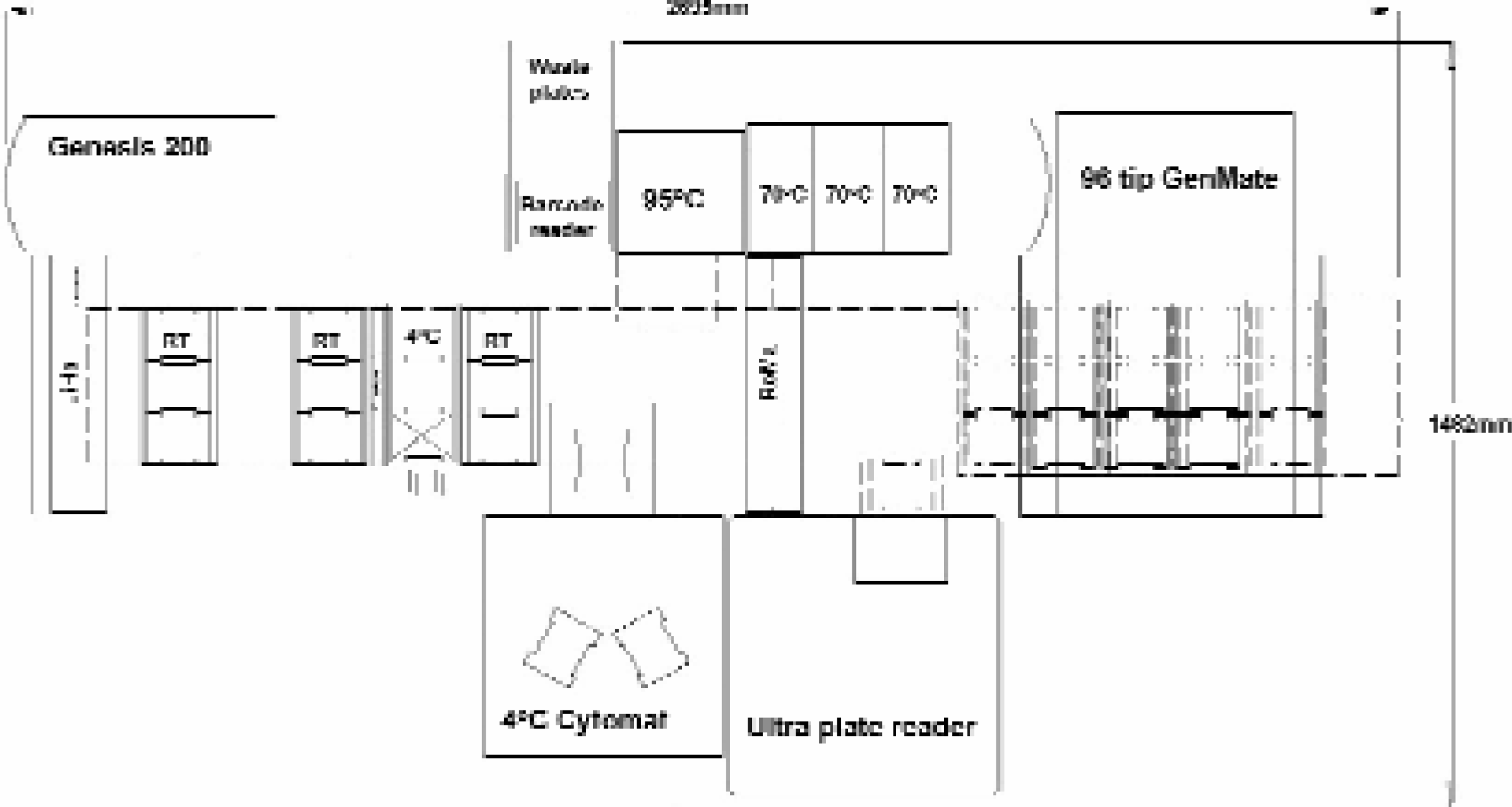
The configuration of modules in the completely automated high throughput system. Components include Genesis RSP 200 workstation (with liquid handling arm, LiHa and robot manipulating arm, RoMa); room temperature carriers (RT); chilled carriers (4°C); waste plate chute; 4°C Cytomat; Tecan Ultra plate reader; barcode reader; 95°C incubators (95°C); 70°C incubators and Tecan GenMate.
The lack of rigidity of the PCR MTPs made exact plate positioning with the Genesis robot manipulator arm (RoMa) very difficult. To obviate this issue, rigid black 384-well polypropylene storage plates (Nunc) were used throughout the whole process. An added advantage of using these U- bottomed plates with wide well diameters was that centrifugation steps were no longer necessary. The flow of samples contained in these plates, through the various processes of the assay, is indicated in figure 6.
Reaction plates preformatted with targets for SNP determination were stored in the random access 4°C humidified Cytomat, together with two 96-deep well plates (DWPs) containing the SNP specific ligation reagents and generic RCA reagents. ABgene Mark II storage plates were employed for both ligation and RCA mixes, due to their high working volume facilitating 24 reaction plates to be processed using a single DWP for the ligation mix.
Upon initiation of the assay, the first reaction MTP containing the DNA targets and the ligation reagent DWP were transferred from the Cytomat storage unit to a customised cooling rack on the Genesis workstation. The cooling rack was custom designed to fit closely the ABgene DWP and to ensure its contents remained at 4°C. This 96 DWP ligation reagent plate contained sufficient quantities of probe to populate 96 384-well MTPs (with up to 96 different SNPs). 5μl of ligation mix was transferred using the 8-tip liquid handling arm (LiHa) of the Genesis workstation. The LiHa possesses eight independently addressable tips that allow the contents of a single source well to be dispensed across the entire 384-well reaction plate. Dispensing of the reagent was executed using pinch valves above the liquid level in the destination well. This strategy was employed to allow rapid multi-dispensing without the possibility of cross-contamination of DNA samples. As with the GenMate, water washes of the Teflon coated Genesis dispensing tips were used to prevent carry over of probes. Due to the reasons outlined previously, automated heat-sealing of plates was not deemed suitable to prevent liquid losses during the incubation steps. Moreover, at the time there were no automated means of removing seals, thereby preventing the use of them in a fully automated process whereby plate reading can be performed without manual intervention. Oil overlay was therefore employed to prevent evaporation losses.
Subsequently, the reaction plate containing the DNA under interrogation with the probe reagent was transferred to the GenMate using the RoMa whereupon 10μl of mineral oil was dispensed into each well. Oil dispensing was performed with 250μl DITIs using slow aspiration and dispense speeds. Dispensing was done above the liquid level onto the side of the well wall. Dispensing above the reaction reagents removed the need for washing the DITIs between subsequent quadrants on a single reaction plate. However, water washing was done between reaction plates to remove residual mineral oil from the surfaces of the DITIs. The reaction plate was then transferred to the 95°C incubator for the denaturation step. The heating platform of the 95°C incubator was custom designed to provide intimate contact with the 384 Nunc reaction plates. Reaction plates were then transferred to the 70°C incubators for ligation of the probes to occur. These incubators were custom modified from Tecan 37°C incubators to reach temperatures of 70°C.
Once the 65°C ligation incubation was completed, the reaction plate was transferred onto the GenMate. The generic RCA reagent DWP was transferred from the Cytomat to a second custom made cooling rack on the GenMate whereupon 10μl of the reagent was dispensed into the reaction wells. A single RCA reagent DWP contains sufficient reagent volume for forty 384-well reaction plates. The liquid handling steps for RCA reagents were identical to those employed in the medium throughput protocol. The reaction was then placed back into the 70°C incubator to enable polymerase activity.
It was observed that the oil overlay did not adversely affect the Ultra's plate reading ability resulting in SNP calling accuracy being unaffected by the presence of the overlay. The overall signal intensity from the wells was reduced. Increasing the amplification from photomultiplier tube by circa 10% compensated for this. Therefore, liquid transfer of reactions from beneath the oil was not required. Furthermore, due to the rigidity of the storage plates used, the reaction plates could be directly read in the Ultra reader without the need for a positioning plate, as was the case with the PCR MTPs. After the fluorescence data for the plate were saved, the plate was discarded through a waste chute mounted at the rear of the Genesis workstation.
Liquid handling scripts were written in Gemini and GenEditor (Tecan) for the Genesis and GenMate modules, respectively. Control and scheduling of all modules were accomplished using FACTS software (Tecan). In addition to device integration, the database within FACTS facilitated tracking and management of samples through the system. A bar code reader was installed on the Genesis workstation and reaction plate identifiers were logged into the FACTS database together with all reagent plates involved in the process, to allow sample traceability.
Simulations based on current protocols indicate that a study of 96 SNPs screened against 96 DNA samples, (9216 SNP calls, i.e., twenty-four 384 well MTPs) would be completed in less than six hours (figure 7). This would involve 24 reaction MTPs, a ligation mix DWP and a RCA mix DWP being loaded into the Cytomat before each batch run. Analysis of this protocol revealed that the LiHa addition of ligation mix is rate limiting. This relatively slow liquid handling step restricts the rate at which reaction plates can be loaded onto the system.
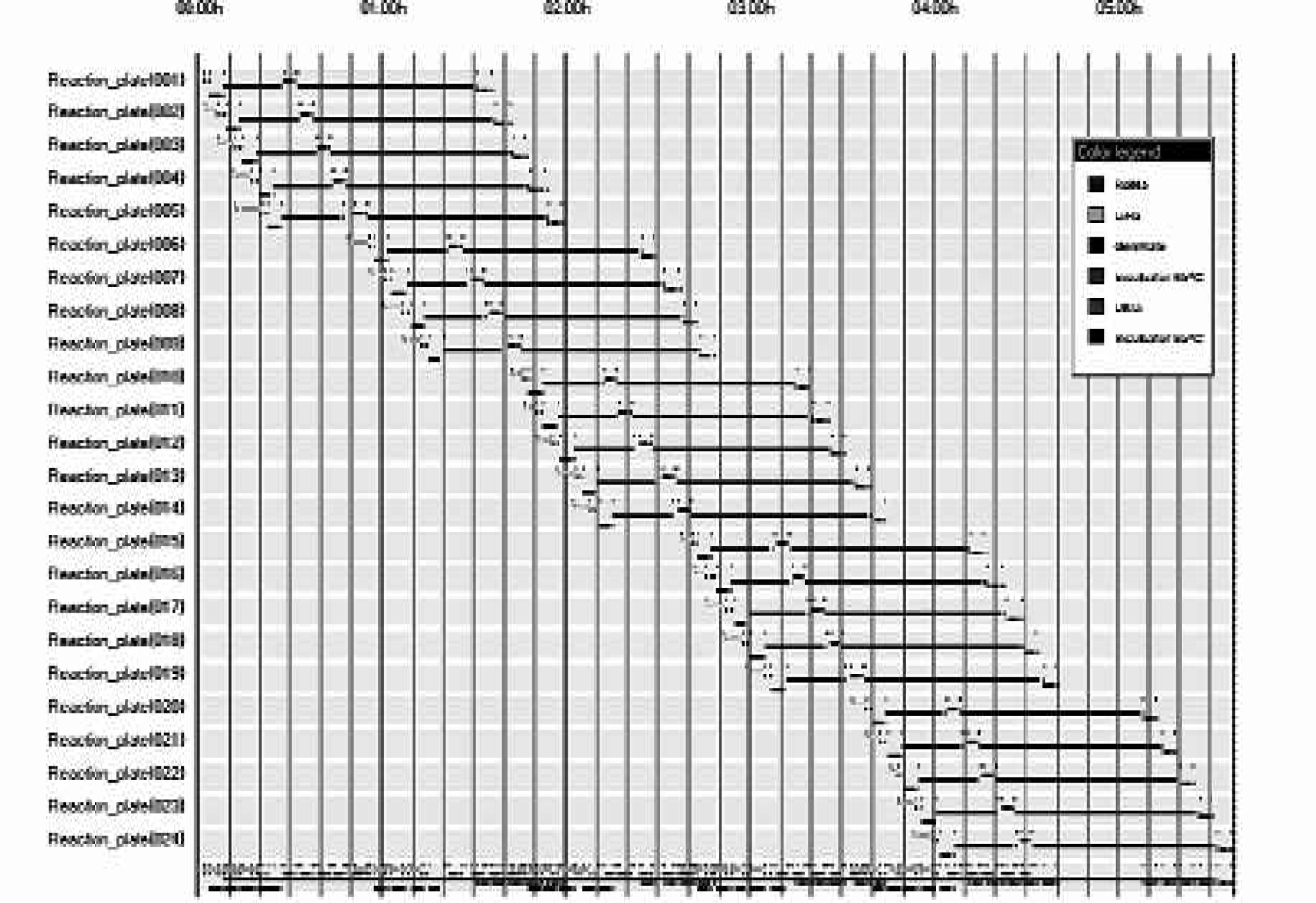
Gantt chart of real time scheduling of the automated process.
Further modeling demonstrated that a larger study of 30,000 SNPs should be possible in a standard working day. This would require reloading the Cytomat 4000 two further times (after all 384-well reactions plates had been processed) leaving the last batch to run overnight.
The system has the capacity to handle 10,000 reactions allowing a batch run of 24 reaction MTPs to be performed without further loading of plates. This can be further increased by use of higher capacity cooled storage devices such as the Cytomat 6000, which has a five-fold greater storage capacity. A greater number of MTPs could be processed without the need for reloading and a throughput of 46,000 SNP calls in a 24-hour period should be possible. Detailed analysis of the throughput predictive modeling reveals that three 70°C incubators (18 incubations sites) are sufficient to eliminate the incubation step as a bottleneck. The single RoMa is consequently rendered rate limiting due to the restricted rate at which MTPs can be transferred between modules. The use of a second RoMa would alleviate this restriction, making higher throughputs possible.
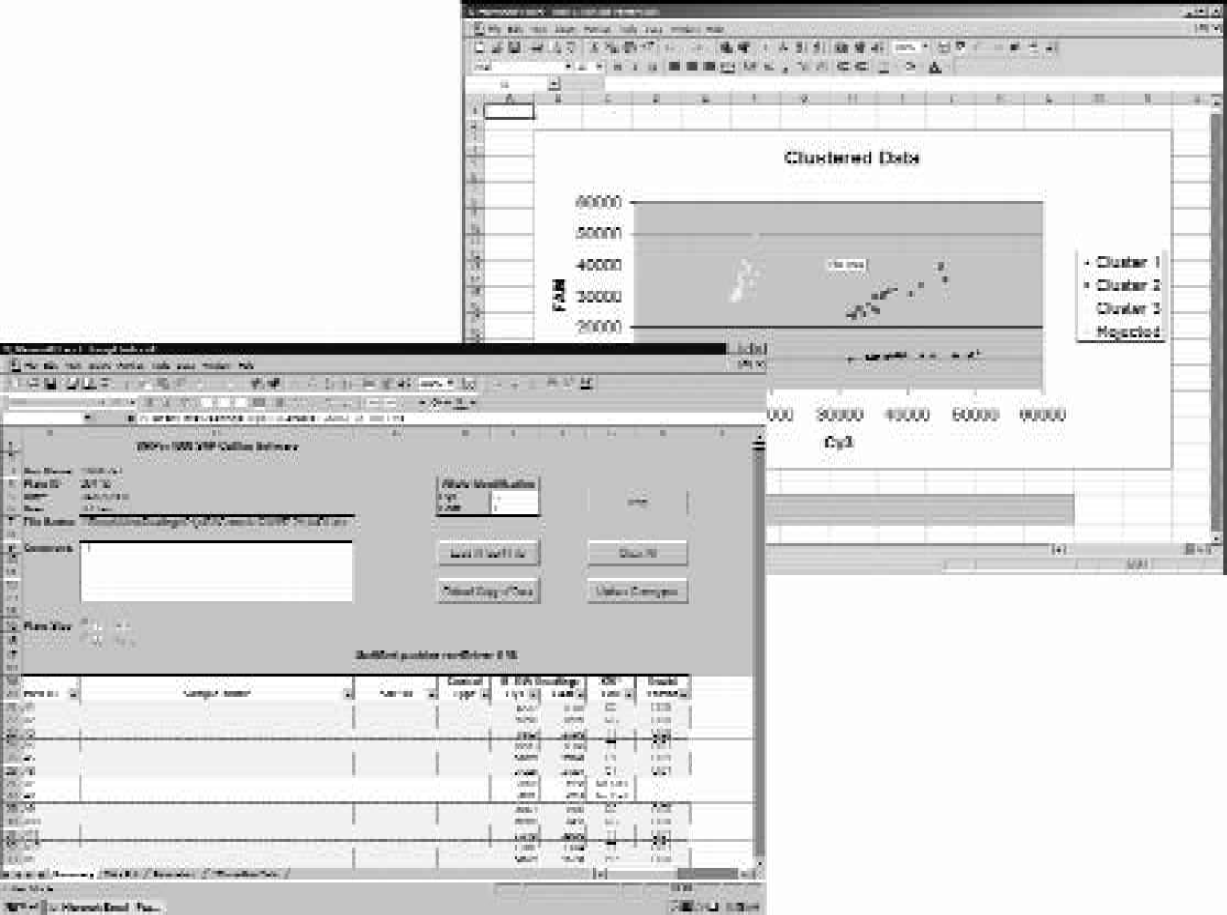
Screen captures of graphic representation and summary table generated by the cluster analysis software.
Data Analysis
The data analysis software used for SNP calling was developed in-house. The requirements were for an algorithm that gave greater than 99% accuracy on SNP calling and which provided quality measures, both for an individual SNP call and for the global data set presented for SNP calling.
The main objective of the data analysis is to partition the data set into classes or clusters, for example, homozygous XX, homozygous YY, or heterozygous XY. In probabilistic cluster analysis, or fuzzy clustering, the probability with which each data point is assigned to a cluster is calculated, and these probabilities, or degrees of membership, can be employed in the provision of the required quality measures.
A number of fuzzy clustering algorithms were evaluated for this application, including the fuzzy c-means algorithm and the Gustafson-Kessel algorithm. 8 The overall best accuracy was obtained from a modified form of the fuzzy c-means algorithm. The data points in this application tend to form linear clusters radiating from the point of origin, rather than point clusters. Clustering in the modified algorithm was based on angular co-ordinates rather than standard Cartesian co-ordinates, and was designed to recognise lines. This was found to be more accurate than the clustering in the standard fuzzy c-means algorithm, which is designed to recognise point clusters.
Quality measures for individual SNP calls were derived from doubt factors defined by the ratio:
(highest degree of membership.)
The doubt factor will be close to zero when the highest degree of membership is much greater than the degree of membership to any other cluster, and close to one for a data point that falls between clusters.
The algorithm was developed and implemented in the MS Excel 97 VBA (Visual Basic for Applications) environment. By developing in this environment, the advantages of in-built MS Excel functionality in the areas of graphical representations, data manipulation and data exporting were exploited. Data were displayed both graphically and within a summary table (figure 7) to facilitate easy and rapid organisation.
Data sets from the manual, semi-automated and fully automated protocols described above were analysed using the fuzzy cluster software (figure 9). Calling accuracy was consistently greater than 99% in all formats. It was, however, apparent that in the fully automated system there were a number of reactions yielding low fluorescence readings resulting in a lack of clear demarcation from the negative control cluster. This may have been a consequence of the difficulties in thermal regulation of the storage plates within the incubators. These plates possess thick polypropylene walls resulting in a high plate heat capacity. Changing reagent temperature within the wells is relatively slow. In the medium throughput protocol, the PCR MTPs utilised have very thin well walls and thus thermal transfer from the heating block to reagents within the well is rapid. Moreover, the thermocyclers, unlike the incubators, exhibit active cooling mechanisms to improve ramping speeds. Modifications aimed at improving ramping speeds in the high throughput system are currently being investigated to optimise reaction performance.
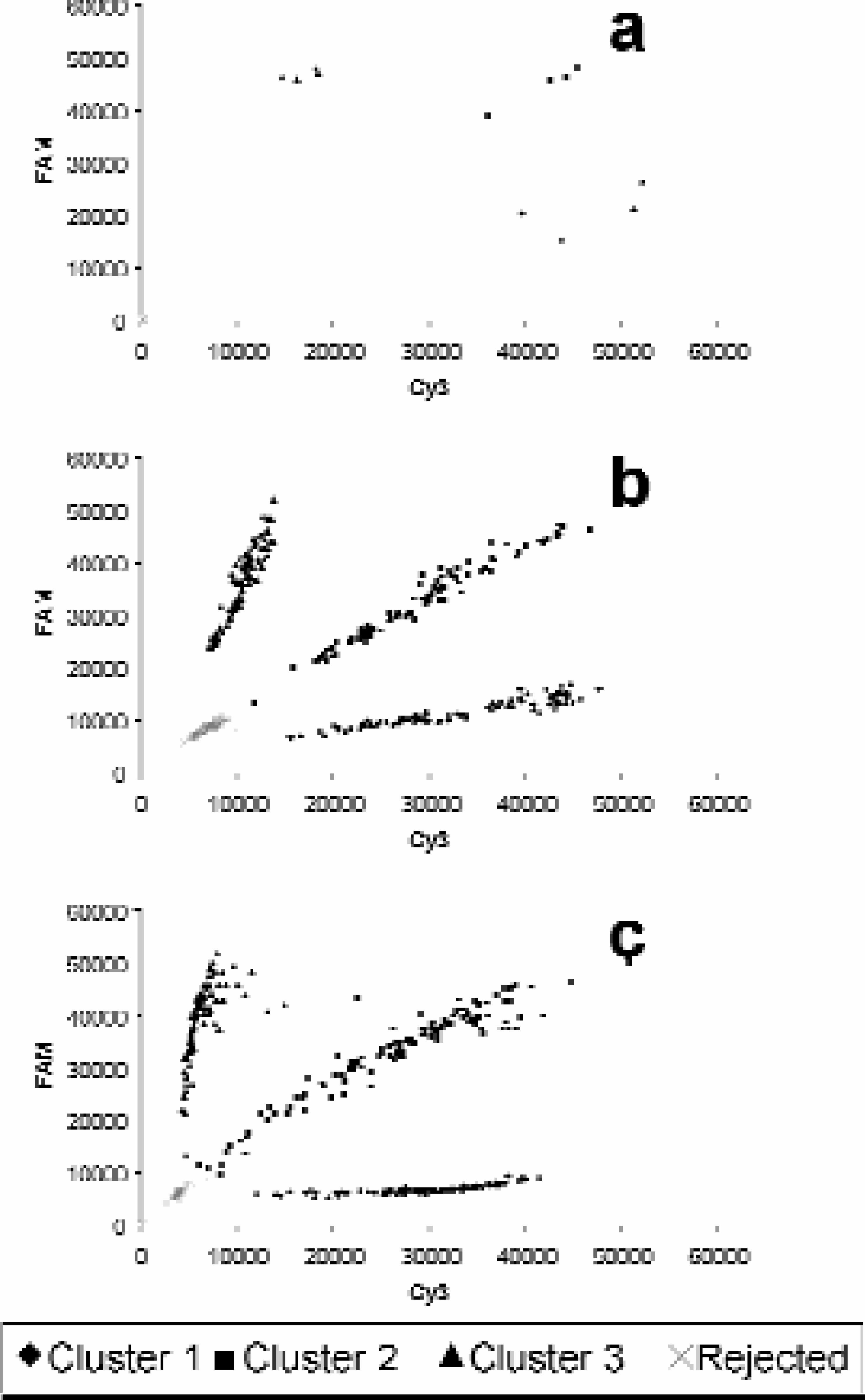
Data points generated by manual, medium throughput and high throughput protocols (a, b and c respectively). Experiments were done using equal numbers of each genotype incorporating an equivalent number of negative controls (i.e. 96 of each genotype and 96 negative reactions). Analysis software identifies clusters and allocates a SNP call, or a rejected call (negative controls) for each data set. Call accuracies for data sets a, b and c were 100%, 100% and 98% respectively.
Conclusions
The systems described above provide scalable solutions to automating an RCA-based SNP calling assay. At the current high throughput levels, the assay can be completely automated with no need for manual intervention after the initial loading of plates. Key automation issues for assay performance were found to have been (1) thermal regulation of reaction wells, (2) evaporation and intraplate liquid migration, (3) cross-contamination during liquid handling steps, and (4) direct fluorescence reading of reaction plates.
Further optimisation of this system is aimed at improving assay performance and increasing throughput. Improvements to the integrated system are presently being investigated. These include:
Reducing reaction volume
Workflow software to manage samples, reagents, probes and data during the SNP scoring process.
Assessment of higher throughput levels by configuring multiple systems working in parallel or using conveyor-based automation.
Footnotes
a
Rolling Circle Amplification (RCA) is covered by patents exclusively controlled by Molecular Staging Inc. A licence to use the RCA process for certain research and development activities accompanies the purchase of certain reagents from Amersham Pharmacia Biotech Limited and affiliates. Amplifluor is a trademark of Intergen Company LP.