
Abstract
A simple and effective method for the preparation of platinum nanoparticles (Pt NPs) grown on amino-functionalized halloysite nanotubes (HNTs) was developed. The nanostructures were synthesized through the functionalization of the HNTs, followed by an in situ approach to generate Pt NPs with diameter of approximately 1.5 nm within the entire HNTs. The synthesis process, composition and morphology of the nanostructures were characterized. The results suggest PtNPs/NH2-HNTs nanostructures with ultrafine PtNPs were successfully synthesized by green chemically-reducing H2PtCl6 without the use of surfactant. The nanostructures exhibit promising catalytic properties for reducing potassium hexacyanoferrate(III) to potassium hexacyanoferrate(II). The presented experiment for novel PtNPs/NH2-HNTs nanostructures is quite simple and environmentally benign, permitting it as a potential application in the future field of catalysts.
1. Introduction
As is well known, platinum nanoparticles (PtNPs) exhibit high catalytic activity for various chemical reactions and have been extensively explored as key catalysts invaluable to many industrial processes [1-5]. In particular, in comparison to bulk platinum, PtNPs not only improve utilization efficiency but also reduce the dosage in catalysis [3]. Nevertheless, the catalytic activity and stability of PtNPs during reactions are still challenges. To overcome these challenges, considerable research effort has been made towards developing novel PtNPs with enhanced catalytic performance.
Immobilization of Pt NPs on catalyst supports has attracted considerable interest. In particular, uniform dispersion and immobilization of the Pt NPs on supporting materials have been actively proven to significantly improve catalytic activity and stability [6-10]. To achieve Pt catalysts with high dispersion, availability, activity, and stability, catalyst supports with developed high surface area, nanoscaled morphology and tuneable surface chemistry are highly desired [2, 11-14].
Halloysite nanotubes (Al2Si2O5(OH)4 2H2O, HNTs), an abundant type of naturally occurring hydrated polymorph silicates with nanotubular structures, are readily obtainable, much cheaper and exhibit interesting features [15]. HNTs consisting of two layered (1:1) aluminosilicates have a predominantly hollow tubular structure, which is regarded to be a potential application as supports for catalytic composites with markedly improved mechanical properties and stability due to a high surface area to volume ratio and different outside and inside chemistry essence [16]. Furthermore, HNTs are approximately 15 nm in lumen size, 50 nm in external diameter and 1000–2000 nm in length [15, 16]. HNTs possess potential anchoring sites for NPs due to the aluminol (Al-OH) groups in the internal surface and Si-OH groups on the external surface [17-19]. Research on noble metal NPs deposited on HNTs have advanced dramatically during the last few years because the as-prepared new functional materials have the desired properties and a large number of applications in different fields [20-24], such as catalysis and fuel cells. Therefore, inspired by their specific structure, HNTs were widely employed as catalyst substrates. However, the efficient attachment of Pt NPs onto HNTs remains a challenge because of the chemical inertness of the HNTs. Thus, it is necessary to modify the surface of HNTs by physical or chemical methods. A common strategy to immobilize metal NPs onto HNTs is the functionalization of their external wall, then following the posterior deposition of the particles [25]. As is known, the presence of functional groups such as -NH2 species on catalyst support surfaces could facilitate the dispersion of metal NPs, resulting in improved catalytic activity [16, 19, 26, 27]. In our previous work, we have introduced a two-step method to synthesize amino-functionalization HNTs (NH2-HNTs) by surface modification with N-(β-Aminoethyl)-γ-aminopropyl trimethoxysilane (APTES) [16, 19]. APTES molecules possess a large number of primary and secondary amino groups with lone electron pairs, which can capture the metal ions to form a chelate complex. Previous research has showed that grafted APTES form a cross-linked network on the surface of HNTs [15]. As reported, the chelate complex exhibits high efficiency for immobilizing NPs onto HNTs [2, 16, 27, 28].
To overcome the above problems, we demonstrate here a novel solution-based strategy for decoration of ultrafine Pt NPs on functionalized HNTs. The synthesis method is simple and effective, allowing various kinds of noble metal NPs uniformly anchored on the surfaces of NH2-HNTs. Among those previous widely employed reducing agents for synthesizing metal NPs such as hydrazine and sodium borohydride (NaBH4), green and renewable materials as the reducing and stabilizing agents are one of the potential methods. Epigallocatechin gallate (EGCG) makes up around 46% of the tea polyphenols (TP), which are groups of water-soluble polyphenol compounds richly deposited in plants [29, 30]. Thus, EGCG was accepted as a green reductant to reduce the metallic ions. In our previous work, we have firstly reported EGCG to synthesize AuNPs successfully [16]. Herein, the green EGCG was introduced as both a reductant and stabilizer to synthesize uniform and well dispersed Pt NPs. It is noteworthy that the synthesis approach does not require the use of undesirable surfactants that may block the active sites on the PtNPs surface, and the obtained Pt nanostructures are stabilized with a layer of EGCG [31–32]. Additionally, a series of characterizations were employed to detect and confirm the nanocomposites. The as-prepared PtNPs/NH2-HNTs nanostructures with ultrafine NPs were utilized as the efficient catalyst material in the reduction reaction and evaluated by the reaction of potassium hexacyanoferrate(III) with NaBH4.
2. Experimental
2.1 Materials
Halloysite nanotubes (HNTs) were obtained by milling and sieving the halloysite clay from Hebei Province (China). Chloroplatinic acid (H2PtCl6 6H2O, 99.9%) was acquired from Shanghai Civil Chemical Technology Co., Ltd. Epigallocatechin gallate (EGCG, 98%) was purchased from Xuancheng BaiCao Plant Industry and Trade Co., Ltd. N-(β-Aminoethyl)-γ-aminopropyl trimethoxysilane (APTES) and sodium borohydride (NaBH4) were supplied from Sinopharm Chemical Reagent Co., Ltd, China. Potassium hexacyanoferrate(III) was supplied by Aladdin Chemistry Co., Ltd. All the chemicals (analytical grade) were used as received without further purification. Deionized water (DIW, 18.2 MΩ) was used to make up all solutions.
2.2 Fabrication of NH2-HNTs nanostructures
The amino-functionalized HNTs (NH2-HNTs) were prepared using a method in our previous work [16]. In a typical synthesis procedure, the nature occurred HNTs was firstly purified by a repeated washing/centrifugation progress and then dried at 80°C in a vacuum oven for 24 hr. A mixture containing 5 g APTES and 95 g 95% ethanol was stirred to get a homogenous solution and then an appropriate amount of acetic acid was added dropwise into the mixture to control the value of pH at about 4. Approximately 5 g of HNTs powder was added, and the suspension was dispersed ultrasonically for 30 min. The suspension was then kept under refluxing conditions for 6 hr at 80°C under constant stirring [25]. The functionalized HNTs underwent an extensive washing/centrifugation process with ethanol to remove excess APTES and possible products of hydrolysis, and then vacuum dried overnight in an oven at 80°C.
2.3 Fabrication of PtNPs/NH2-HNTs nanostructures
100 mg functionalized HNTs were dispersed in 50 mL of deionized water without surfactants and stirred for 1 hr to obtain a homogenous solution. After being stirred, the mixture was ultrasonically dispersed at room temperature for 1 hr. Then H2PtCl6 6H2O was dissolved in deionized water to obtained a 10 mM Pt4+ solution; 10 mL Pt4+ solution was added into the mixture, and then the mixture was heated to 60°C and stirred by magnetic agitator for 2 hr. At last, 30 mg EGCG dissolved in 5 mL deionized water was dropped in the mixture and stirred vigorously for 12 hr until a brown colour was observed, indicating the formation of metallic PtNPs on the surface of functionalized HNTs. The as-prepared powder was filtered, extensively purified through a repeated washing/centrifugation process with ethanol and deionized water to remove the excess EGCG, and then dried in an oven at 60°C overnight under vacuum and used for further experiments and characterizations.
2.4 Characterizations
A Nicolet 5700 Fourier transform infrared (FTIR) spectrometer was used to record the FTIR spectra of the samples in the range of 4000–400 cm−1. The samples were individually spread on KBr discs and then dried under an infrared lamp. The X-ray diffraction (XRD) patterns of pristine HNTs and PtNPs/NH2-HNTs were collected on a SIEMENS Diffraktometer D5000 X-ray diffractometer using Cu Kα radiation source at 35 kV, with a scan rate of 0.02° 2θ s−1 in the 2θ range of 10–90°. The microscopic features of the samples were observed with a field-emission scanning electron microscope (FE-SEM) (JSM-6700F, JEOL Japan) at an acceleration voltage of 3 kV. Element content was analysed using an energy dispersive spectrometer (EDS) at 10 kV with FE-SEM. The number of fringes and lateral dimensional of Pt NPs (100 counts) were measured with Image-Pro Plus 6.2 software. TEM images and SEAD patterns of all the samples were observed with a transmission electron microscope (TEM) (JSM-2100, JEOL Japan) at an acceleration voltage of 200 kV. The surface chemical composition of pristine HNTs, NH2-HNTs, and PtNPs/NH2-HNTs were testified by X-ray photoelectron spectrometer (Kratos Axis Ultra DLD) with an aluminium (mono) Kα source (1486.6 eV). The aluminium Kα source was operated at 15 kV and 10 mA. The catalytic reduction reaction was recorded at the absorption band 320–500 nm in the UV-vis spectra using a U-3010 UV-vis spectrophotometer (Hitachi).
2.5 Catalytic Activity Measurements
The catalytic activity of the samples was evaluated by the reduction of potassium hexacyanoferrate(III) under 25°C. 0.3 mg PtNPs/NH2-HNTs was charged into 100 mL of a potassium hexacyanoferrate(III) solution (0.27 mM), and the mixture underwent ultrasonic agitation for 30 min at 25°C. Subsequently, an excess of NaBH4 was added to the above solution. Immediately after adding the NaBH4, the absorption spectra were recorded in the range of 320–500 nm by UV-vis spectrophotometer with an interval of several seconds at 25°C. The yellow suspension became colourless within 150 s. It is noted that all solutions were previously deaerated, and the reaction mixtures were maintained at pH 12 to avoid NaBH4 decomposition [33]. For comparison, a similar measurement was also performed without PtNPs/NH2-HNTs catalysts and Pt NPs catalyst in solution only.
3. Results and discussion
3.1 Synthesis of PtNPs/NH2-HNTs nanostructures
Herein, we demonstrate the systematic sketch of the fabrication process of PtNPs/NH2-HNTs nanostructures. The whole preparation strategy is shown in Scheme 1 (for detailed experimental steps, please see Experimental Section). In the first step, the pristine HNTs were functionalized and individually dispersed in ethanol using APTES to graft amino groups onto the surface of HNTs. It has been proven that the presence of a large amount of -NH2 groups with one lone electron pair could lead to the formation of a chelating complex with Pt ions [16, 23]. In particular, – NH2 groups with positive charge can anchor the Pt ions with a negative charge through coordinate bonds [27, 34, 35] (Scheme 1b). Therefore, the Pt ions are preferable to combine with -NH2 groups, resulting in the formation of PtNPs-NH2 complexes, as will be testified and discussed later. In other words, with the numerous nucleation sites provided by the amino-functionalization HNTs, the Pt NPs were uniformly grown and well dispersed on the surface of the HNTs without agglomeration. In the second step, with the addition of EGCG, thereby introducing multiple phenolic hydroxyls, the previously-formed Pt-NH2 complexes can also chelate with the phenolic hydroxyls of EGCG, together with the reduction of Pt ions to metal Pt by EGCG. Accompanying the reduction of the platinum precursor, the phenolic hydroxyls of EGCG are also oxidized to orthoquinone [16, 36]. In the present process, EGCG served as both a reductant and stabilizer to prevent the Pt NPs from aggregation [16]. Additionally, it should be noted that the HNTs decorated by APTES show high stability in water and ethanol in experiment, which can be probably ascribed to the strong interactions between -OH and APTES molecules.

Schematic presentation of the growth mechanism and the formation process of PtNPs/NH2-HNTs.
3.2 Characterization of PtNPs/NH2-HNTs nanostructures
FTIR was performed to justify the chemical modification of pristine HNTs by APTES and the chemical changes during the functionalization process. Fig. 1 shows the FTIR spectra of pristine HNTs powders (black curve), the functionalized HNTs powders (blue curve), and PtNPs/NH2-HNTs powders (red curve). As can be seen from Fig. 1, the characteristic peaks of pristine HNTs displayed at 3622 cm−1 and 3698 cm−1 are assigned to the O-H stretching vibration of inner and outer -OH groups (between the interface of the Si-O tetrahedron and the Al-O octahedron), respectively [16, 27]. The HNTs display a broad band centred at 1638 cm−1 ascribed to deformation of water. The strong absorption around 1032 cm−1 is assigned to the Si-O stretching vibration in HNTs. The other three characteristic peaks at 912, 550 and 471 cm−1 correspond to the O-H deformation vibration of inner Al-OH groups, the Al-O tetrahedral sheets of deformation vibration of Al-O-Si and the Si-O tetrahedral sheets of deformation vibration of Si-O-Si, respectively. All of the characteristic peaks of HNTs nearly have no change in the functionalized HNTs, indicating that the basic structure of HNTs and functionalized HNTs remain constant [16]. It is noted that new absorption peaks at 2925, 2854 and 1381 cm−1 appeared, which are assigned to the C-H asymmetric and symmetric stretching vibrations of APTES, respectively [23]. Furthermore, the newly emerged absorption peak at 1105 cm−1 ascribed to the in-plane stretching of Si-O indicates that a small distortion of the Si-O structure occurred, confirming the existence of APTES on modified HNTs [37, 38]. Thus, The FTIR results confirm that the APTES has successfully been grafted onto the surface of HNTs.
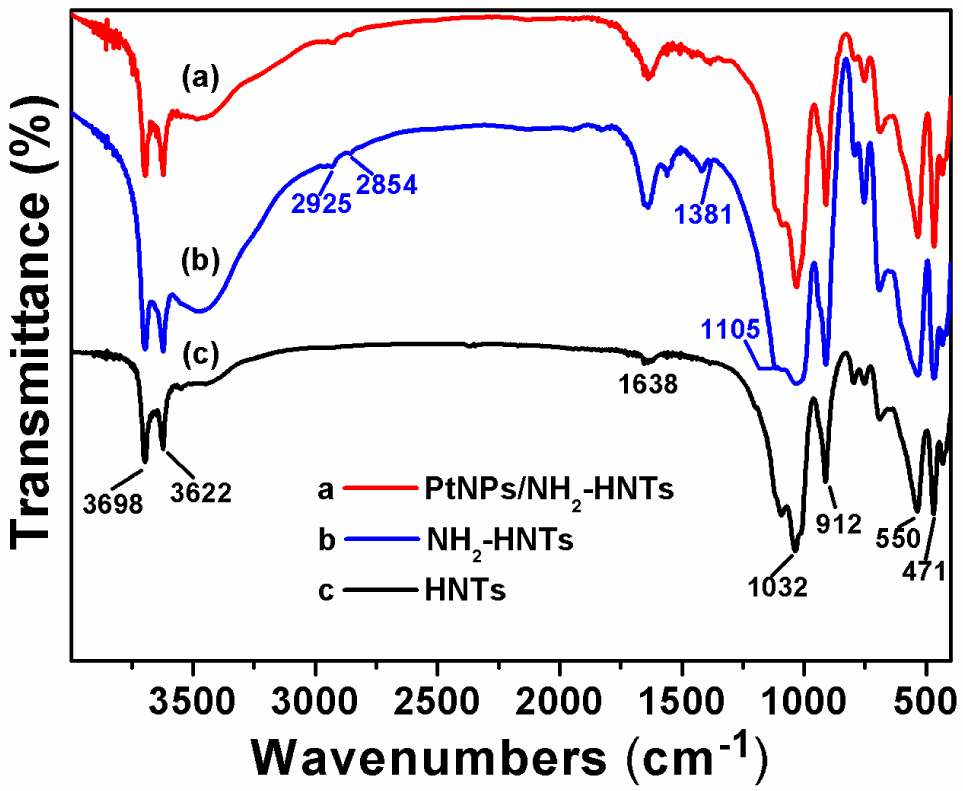
FTIR spectra of pristine HNTs, NH2-HNTs and PtNPs/NH2-HNTs
Fig. 2 shows the SEM images of pristine HNTs and PtNPs/NH2-HNTs under different magnifications. The pristine HNTs possess a fibrous structure and the length of the pristine HNTs ranges from 400–2000 nm (Fig. 2a, b), indicating the polydispersity of HNTs in length. The energy dispersive spectrometer (EDS) pattern of the NH2-HNTs in Fig. 2c shows that the pristine HNTs were composed of the elements of Al, Si and O. After the functionalization of HNTs by APTES, the fibrous structure of pristine HNTs has been retained (Fig. 2d, e). Pt NPs could not be observed on the surface of HNTs under the present magnification due to very small particle sizes. However, according to the EDS result (Fig. 2f) of the PtNPs/NH2-HNTs, the peak of element Pt is newly emerged, demonstrating the successful immobilization of Pt NPs on the PtNPs/NH2-HNTs nanostructure.

FE-SEM images of (a, b) pristine HNTs; (c) the EDS spectrum of the pristine HNTs; (d, e) the FE-SEM images of the PtNPs/NH2-HNTs nanostructures; (f) EDS spectrum of the PtNPs/NH2-HNTs nanostructures
TEM was further used to determine the details of the detailed microstructure of the samples. The images of pristine HNTs (Fig. 3a) reveal a typical tubular structure with an external diameter of approximately 75 nm. As shown in Fig. 3a and inset, numerous ultrafine Pt NPs are found to have grown over the entire NH2-HNTs. In the present investigations, Pt catalysts supported on NH2-HNTs were synthesized by a facile and green approach using epigallocatechin gallate (EGCG) as the reductant [16]. As shown in Fig. 3b, c, Pt NPs are well dispersed with nearly no aggregations, which was mainly due to the stabilization of EGCG [16, 36, 39–40]. Fig. 3b also shows that there is essentially no obvious change in the size and structure of the HNTs during the growth of Pt NPs on its surface. The size distribution histogram of the Pt NPs (Fig. 3d) was calculated from the corresponding HRTEM image in Fig. 3c. The particle size distribution was created by measuring 100 particles and the average particle size of the Pt NPs was only around 1.5 nm.

TEM images of (a) pristine HNTs, and (b, c) PtNPs/NH2-HNTs nanostructures, (d) The corresponding average diameters of Pt NPs immobilized on NH2-HNTs
As shown in Fig. 4a, Pt NPs nearly no aggregations and densely dispersed on the NH2-HNTs are observed. The crystallographic orientation of the Pt NPs was investigated by HRTEM, which is taken on the area marked in the rectangle in Fig. 4a. Crystalline fringes of Pt NPs were observed under close inspection of the Pt NPs from HRTEM. The closely packed Pt NPs contain single-crystal atomic structures, growing in the <111> direction, with a lattice spacing of about 0.23 nm, which corresponds to the lattice spacing of the (111) planes of bulk Pt crystal [41-45]. It can be seen in Fig. 4c that the selected-area electron diffraction (SAED) pattern reveals two concentric rings, attributable to the (111) and (200) crystal planes of fcc Pt crystal. The SAED pattern further indicates that the Pt NPs is polycrystalline and the diffuse ring is attributed to the existence of EGCG [16]. Subsequently, the successful synthesis of the Pt catalyst loaded onto the HNTs could be further substantiated by XRD. Fig. 4d shows the typical XRD patterns of HNTs before (red line) and after (black line) the anchor of Pt NPs. According to the literature, all of the emerged peaks can be indexed to the characteristic peaks of pristine HNTs [16, 22], as shown in curve 1. In Fig. 4d (black line), although the intensity is quite weak compared with those of HNTs, three relative diffraction peaks at 39.7°, 46.2°, and 67.4° are still apparent after the formation of Pt NPs, which match exactly the reference values for the typical (111), (200) and (220) reflections corresponding to pure Pt crystals with a face-centred cubic (fcc) structure, according to previous reports [45, 46], in good agreement with the SAED analysis in Fig. 4c.

(a) Low-magnification TEM image of PtNPs/NH2-HNTs nanostructures. (b) High-resolution TEM (HRTEM) image of Pt NPs taken on the area marked in the rectangle in (a). (c) SAED pattern of the Pt NPs shown in (b). (d) Typical XRD patterns of the pure HNTs powder (curve 1) and PtNPs/NH2-HNTs powder (curve 2). (h: HNTs).
To investigate the chemical states of the surfaces of PtNPs/NH2-HNTs, XPS analysis of the pristine HNTs powder, NH2-HNTs powder, and PtNPs/NH2-HNTs powder were carried out. As depicted in Fig. 5a, overlapping peaks of Pt 4f and Al 2p spectra from PtNPs/NH2-HNTs were observed. For Pt 4f, the spectrum consists of doubled components (4f7/2 and 4f5/2). Peaks at 71.57 eV (4f7/2 components) and 74.4 eV (4f5/2 components) correspond to metallic Pt, implying that PtNPs are successfully synthesized on the surfaces of the amino-functionalization HNTs. Meanwhile, the higher-binding-energy components in fitting curves demonstrated the content of Al-OH at 73.39 eV and the Al-O at 75.6 eV [27]. As shown in Fig. 5b, the intense peak located at 531.8 eV corresponds well with O 1s of pristine HNTs (black curve). In the meantime, the O 1s peak of PtNPs/NH2-HNTs (red curve) fit into two individual component peaks labelled 532.0 eV and 532.5 eV. Compared with the pristine HNTs, it is apparent that the observed binding energy at around 532.5 eV of PtNPs/NH2-HNTs may be related to O 1s from APTES, and the low shift of O 1s binding energy from 531.8 eV to 532.0 eV for PtNPs/NH2-HNTs may be due to a coupling reaction between HNTs and amino groups, implying that the – NH2 was successfully grafted on the HNTs [27]. Fig. 5c shows the Si 2p XPS spectrum of the HNTs and PtNPs/NH2-HNTs. Similar to the binding energy of O1s, after HNTs being modified by amino groups and then immobilized by Pt NPs, the Si 2p spectrum of PtNPs/NH2-HNTs exhibits a peak at 102.6 eV, which is ascribed to the Si-OH of HNTs, in the meantime, the other peak with binding energy at 103.2 eV represents the Si-O of APTES [25]. The changed binding energy for Si-OH and the newly emerged binding energy for Si-O might be due to a coupling reaction between HNTs and amino groups. To further verify the chelating effects of the N atoms in NH2-HNTs with Pt atoms, the XPS survey of N atoms for NH2-HNTs and PtNPs/NH2-HNTs were performed. As observed in Fig. 5d, the peaks at around 399.4 eV and 401.5 eV are attributed to the two types of N atoms (-NH2 and -NH-) in APTES grafted on the HNTs. Furthermore, Fig. 5e presents the N 1s spectrum of obtained PtNPs/NH2-HNTs. The doubled peaks that emerged at 400.1eV and 401.6 eV are in agreement with the two chemical states of nitrogen in the PtNPs/NH2-HNTs [16]. Notably, after the nucleation of Pt nanoparticles, the low shift of the N 1s binding energy for PtNPs/NH2-HNTs is attributed to the chelating interactions of the Pt NPs with the N atoms in the NH2-HNTs. Meanwhile, N 1s peaks for the PtNPs/NH2-HNTs confirm that APTES has been anchored onto HNTs. Depending on the aforementioned analysis, these results verify that the -NH2 species have been successfully grafted onto HNTs and the chelating effects between -NH2 and Pt NPs.
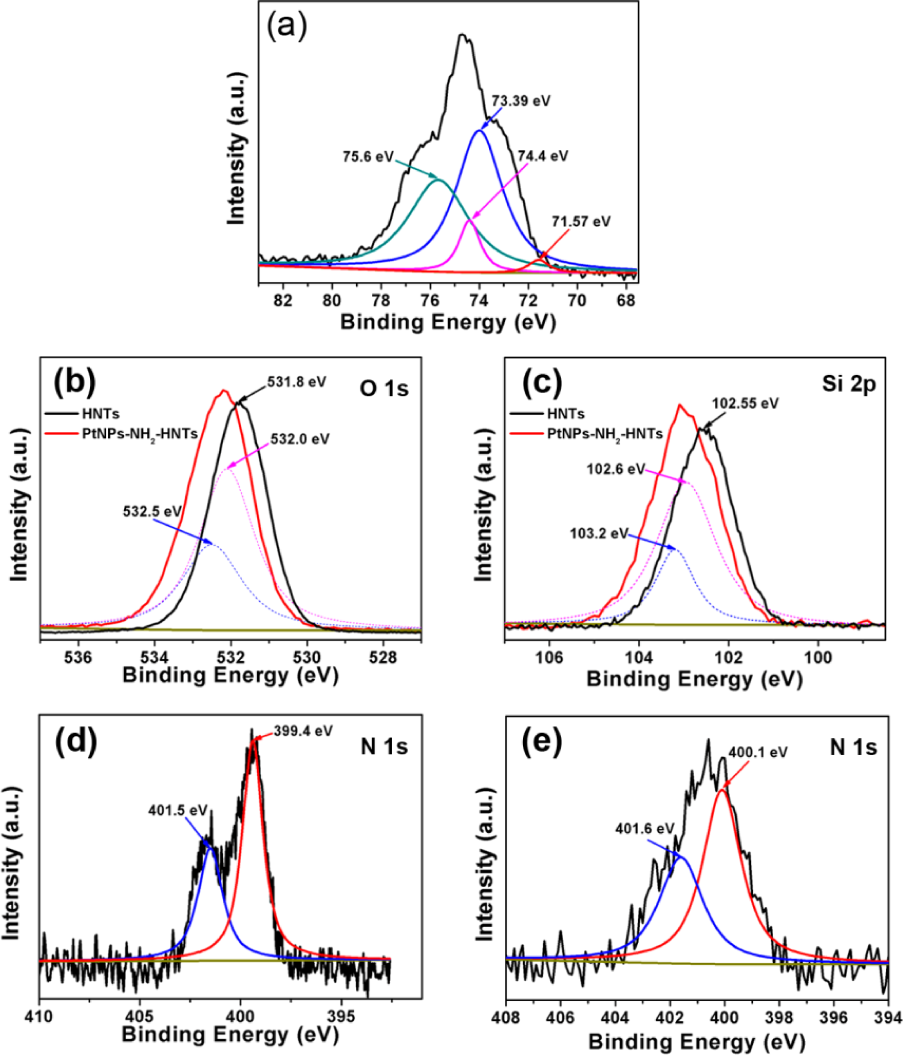
XPS survey of (a) Pt 4f and Al 2p XPS spectra of PtNPs/NH2-HNTs; (b, c) O 1s and Si 2p XPS spectra of pristine HNTs and PtNPs/NH2-HNTs; (d, e) N 1s XPS spectra of NH2-HNTs and PtNPs/NH2-HNTs
3.3 Catalysis investigations of PtNPs/NH2-HNTs nanostructures
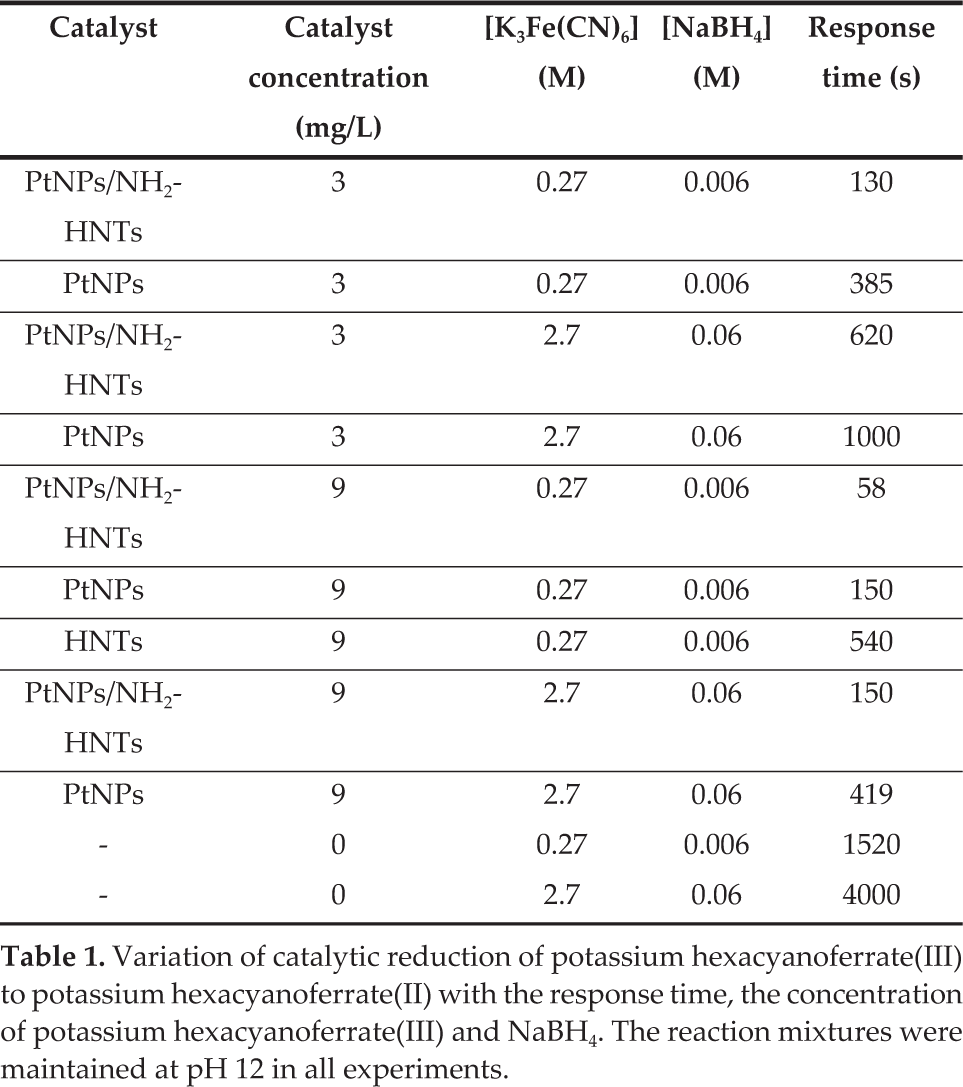
As described above, we have fabricated the PtNPs/NH2-HNTs nanocomposites successfully. High dispersion of ultrafine PtNPs can be useful for many potential applications. Considering the uniform, good dispersion and excellent stability of the prepared PtNPs/NH2-HNTs nanostructures, we were inspired to exploit them as a remarkable catalyst. Herein, we employed a reaction of reducing potassium hexacyanoferrate(III) by borohydride ions in aqueous solution to investigate the catalytic properties of the Pt nanostructure [5, 47], which has been proposed as a model electron-transfer reaction to evaluate the catalytic activity of noble metal nanoparticles [48]. In addition, it is worth mentioning that, compared with the catalyst-free reaction, the reactions between potassium hexacyanoferrate(III) and borohydride ions have been confirmed to be efficiently catalysed onto noble metal nanoparticles, such as gold and platinum nanoparticles [47]. Furthermore, the reaction was previously reported to behave as a zero-order reaction without metal nanoparticles, whereas in the presence of noble metal nanoparticles the reaction mechanism becomes first order [48]. The catalytic efficiency was thus evaluated by measuring the effect of the PtNPs/NH2-HNTs on the reduction rate of potassium hexacyanoferrate(III) by NaBH4. Here, the kinetics of the reduction process can not be treated as a model electron-transfer reaction but a pseudo-first-order reaction due to the concentration of NaBH4 was excessive in all experiments [47]. Moreover, the process of the reduction was monitored through changes in the UV/Vis spectra, as shown in Fig. 6 for different concentrations of potassium hexacyanoferrate(III) and catalyst. Fig. 6 shows that the characteristic absorption peak of potassium hexacyanoferrate(III) at 420 nm decreases with time as it is reduced to potassium hexacyanoferrate(II) for various concentrations of catalyst and potassium hexacyanoferrate(III). The observed spectral changes were also reflected in the colour changes of the solution during the reaction (inset in Fig.6 a, b). Meanwhile, with addition of PtNPs/NH2-HNTs catalyst, the typical yellow colour for potassium hexacyanoferrate(III) gradually disappeared. Notably, the reduction time for 0.27 mM and 2.7 mM of potassium hexacyanoferrate(III) is less than 130 s and 150 s, respectively (Table 1). A similar catalytic effect was observed with Pt NPs in solution only. It is also noted that the catalysed reaction times for 0.27 mM and 2.7 mM are slow after the addition of the Pt catalysts, which are 385 s and 419 s, respectively (Table 1). As seen in the insets of Fig. 6, the kinetic data were roughly fitted with the integrated first-order kinetics of equation (1):
Variation of catalytic reduction of potassium hexacyanoferrate(III) to potassium hexacyanoferrate(II) with the response time, the concentration of potassium hexacyanoferrate(III) and NaBH4. The reaction mixtures were maintained at pH 12 in all experiments.

Successive UV-vis spectral evolution of potassium hexacyanoferrate(III) in the presence of borohydride (NaBH4) and catalyst: (a) [K3Fe(CN)6]=0.27 mM, [PtNPs/NH2-HNTs]=3 mg/L, [NaBH4]= 0.006 M, pH= 12; (b) [K3Fe(CN)6]=2.7 mM, [PtNPs-HNTs]=9 mg/L, [NaBH4]= 0.06 M, pH= 12; (c) [K3Fe(CN)6]=0.27 mM, [PtNPs]=3 mg/L, [NaBH4]= 0.006 M, pH= 12; (d) [K3Fe(CN)6]=2.7 mM, [PtNPs]=9 mg/L, [NaBH4]= 0.06 M, pH= 12. Inset (Fig. 6a, b) shows optical images of the corresponding solutions. The insets show the linearized data of the experimental results to first-order analysis according to integrated kinetic equation (1) for the corresponding data to the Fig. 6.

The kobs of these reactions for PtNPs/NH2-HNTs and Pt NPs in the variation of catalytic conditions are calculated to be 0.0268, 0.0107, 0.0088, and 0.0061 s−1, respectively. In the same chemical reaction addition, the values are comparable to those previously reported for Pt NPs supported on other catalyst substrates [47].
The catalytic effectiveness of variations of catalyst and without any catalyst were tested by the reduction of potassium hexacyanoferrate(III) in room temperature, which showed the high catalytic effectiveness of the PtNPs/NH2-HNTs (Table 1). However, without a catalyst, the reduction process was observed slowly. As shown in Table 1, with the incorporation of catalysts the response time of potassium hexacyanoferrate(III) significantly decreased and the reaction mechanism became first order (Fig. 6). Besides, when increasing the amount of catalyst, the reaction process can be further significantly reduced. Furthermore, it can be seen that the Pt NPs still possess high catalytic activities after being anchored on amino-functionalized HNTs. However, compared with PtNPs/NH2-HNTs and Pt NPs in solution, the reduction time for 0.27 mM of potassium hexacyanoferrate(III) was more than 540 s with only HNTs, implying that the results were mainly due to Pt NPs. To further investigate the effectiveness of catalyst supports, we performed an experiment to test Pt NPs only in a solution catalyst by reducing the potassium hexacyanoferrate(III) in the same conditions (Table 1). As shown in Table 1, the catalytic effectiveness of Pt NPs in solution is relatively lower than that of the PtNPs/NH2-HNTs catalyst. The presented experiment for the novel PtNPs/NH2-HNTs heterostructures are quite simple and environmentally benign, permitting it to be a potential application in the future field of catalysts. In addition, the PtNPs/NH2-HNTs heterostructures could be easily separated from the reaction system by filtration or centrifugation and reused, demonstrating the advantages of using HNTs as the catalyst support. Notably, the synthesis method here provides a blueprint for designing various noble metallic NPs anchors on hollow structures.
4. Conclusion
In summary, we developed a simple and effective method for the preparation of Pt NPs grown on amino-functionalization HNTs. The results show that PtNPs/NH2-HNTs nanostructures with ultrafine Pt NPs were successfully synthesized by green chemically-reducing H2PtCl6 6H2O without using concentration surfactant. The as-obtained nanostructures were utilized as the efficient catalyst material for the reduction reaction, exhibiting promising catalytic properties for reducing potassium hexacyanoferrate(III) to potassium hexacyanoferrate(II). The presented experiment for the novel PtNPs/NH2-HNTs nanostructures is quite simple and environmentally benign, permitting it to be a potential application in the future field of catalysts.
Footnotes
5. Acknowledgements
We acknowledge the support of the project of the National Natural Science Foundation of China (NSFC) (51373154), the 521 Talent Project of Zhejiang Sci-Tech University.