
Abstract
In the current review, we discuss the process of modeling pediatric epileptic encephalopathies with a focus on in vitro iPSC-based technologies. We highlight the potential benefits as well as the challenges of these approaches and propose appropriate standards for the field.
Pediatric Epilepsy and Genetic Origins
Epilepsies are diseases of episodically dysfunctional neuronal circuit activity in the brain (1). Epilepsy is currently estimated to affect 3.4 million Americans and over 65 million individuals worldwide, constituting one of the most prevalent neurological diseases (2, 3). While some types of epilepsies present with mild seizures occurring early on in neonates or infants, others present with severe epileptic encephalopathies (EE) with intellectual disability, pharmacoresistance and other multi-organ comorbidities (for in-depth reviews on clinical seizure classifications, see [1, 4]). Severe EEs of infancy and childhood are a group of devastating diseases, characterized by frequent seizures and abundant epileptiform activity on EEG (4). EEs are associated with developmental delay or regression that might be preexisting or follow seizure onset and exacerbation, respectively. Frequent comorbidities include behavioral and movement disorders, with often poor outcomes (4). Genetic screening in epilepsy patients, widely enabled by the advent of next-generation sequencing (NGS), has allowed for the identification of many monogenic missense, nonsense, and truncation mutations causing maladaptive changes in the function or expression of specific proteins (Figure 1). Currently, the Online Mendelian Inheritance in Man (OMIM) database lists over 2,277 entities when the search term “epilepsy OR seizure” is used. The detection of mutations has dramatically advanced our understanding of the underlying pathophysiology in idiopathic epilepsy in the last 20 years. The percentage of EE cases believed to result from specific genetic mutations (currently over 70%) continues to increase as more genetic sequences become available (for in-depth reviews on the genetics of epilepsy, see [4, 5]). Understanding how genetic mutations in a specific subset of genes contribute to the pathogenesis of early-onset childhood epilepsy during critical stages of brain development has great importance for diagnosis, treatment, and prevention of epilepsy.

Number of mutations identified in epilepsy genes by gene function. One hundred twenty-one epilepsy-associated genes were compiled from the EGI epilepsy gene list (https://www.cureepilepsy.org/egi/genes.asp, see [5, 15, 50]). Genes were categorized using GO slim biological function terms and the number of mutations identified in a gene was compiled from querying HGMD professional database.
Model Systems: All Models Are Useful, No Model is Perfect
Various model systems encompassing both in vitro and in vivo approaches have been successfully used over the years to study genetic epilepsies (for a detailed review on model systems, see [6]). Each model system offers advantages as well as disadvantages and is uniquely suited to addressing a specific set of questions. For instance, heterologous expression systems in non-neuronal cell lines (such as HEK293, CHO, or Xenopus laevis oocytes) can be used to investigate the protein dysfunction caused by a genetic variant or a dominant-negative effect that the mutant protein may exert on the available wild-type counterparts.
For ion channels mutations, comprising nearly half of the mutations identified (Figure 1), these systems provide a high-throughput platform for assessing the biophysical pathogenicity of specific variants using classic or more recently established high-throughput automated patch-clamp techniques such as the SyncroPatch (7). Heterologous expression is a powerful and cost-efficient approach used to screen through genetic variants and establish a genotype–phenotype correlation. However, a protein can function differently in a non-native environment due to disparities in splicing patterns, posttranslational regulation, cellular and protein trafficking processes, interactions with other proteins, and other uniquely neuronal factors. Therefore, the effect of a disease-associated genetic variant on inherent protein function in native neurons may not be fully recapitulated in a heterologous expression system.
More sophisticated whole organism in vivo models engineered with the genetic variation of interest can address effects on intrinsic neuronal and network function, as well as temporal pathogenesis. Drosophila melanogaster models of epilepsy offer the advantage of cost effective and high-throughput genetic interaction studies (8, 9). Genetic epilepsies such as KCNQ2-EE (10) and SCN1A-Dravet Syndrome (11) have also been modeled in zebrafish. These display robust electrical seizure events that can be recorded through EEGs and video monitoring, can support high-throughput mutagenesis as well as antiepileptic drug (AED) screens (12). Mammalian models, including mice, have been widely utilized to study epilepsy mechanisms, as well as in preclinical trials of AEDs (6). These models include animals with spontaneously occurring mutations resulting in an epileptic phenotype, those engineered with disease mutations detected in patients, and models of chronic epilepsy or acute seizures induced by different stimuli (12).
Nonetheless, critical differences in gene expression, protein function, and network participation exist between mice and humans with roughly 20% of CNS genes showing distinct expression patterns in the cortex (13). These differences may account for the disparate efficiencies of AEDs in clinical trials and our inability to adequately treat 30% of epilepsy patients. Thus, there is a requirement for neuronal models that better recapitulate human conditions. The discovery of human induced pluripotent stem cells (iPSCs [14]) that can be readily generated by reprogramming adult somatic cell types, such as skin-derived fibroblasts or blood mononuclear cells, have opened the door to elucidating human pathogenic mechanisms of genetic diseases such as epileptic channelopathies. The expectation has been that iPSC models of epilepsy will bridge the gap between functional studies in heterologous expression systems, animal models, and human clinical presentation of epileptic disorders. To address the cross-correlation of findings among the different models—that is, the functional effects of mutations or the response to pharmacological treatments—it would be interesting to take a top-down approach and study the same genetic variants across a mouse model, a heterologous expression system, and iPSC-derived patient neurons.
iPSC Models: How Do We Standardize and What is the Right Control?
Patients with different mutations in the same gene can have drastically different types and severity of seizures, as well as different clinical responses to pharmacological treatment (15). Studying patient variability with animal models is very difficult; thus, it has been hard to create patient-specific disease models. The unique advantage of iPSCs is that they allow for the study of human disease in the context of each person's unique genetic constellation, in disease-relevant cellular subtypes, as well as providing a platform for examining the effects of disease-associated genetic variants in early developmental stages (Figure 2). Patient iPSCs have been used to effectively model several neurodevelopmental diseases associated with epilepsy including Dravet Syndrome (16–22), Angelman Syndrome (23, 24), neurodevelopmental delay associated with cyclin-dependent kinase-like 5 (CDKL5) gene mutations (25), STXBP1-associated EE (26), and Timothy Syndrome (27, 28); for in-depth reviews on specific findings of iPSC-based epilepsy models, see Tidball and Parent (29). These studies have provided some novel mechanistic insights, including compensatory neuronal mechanisms to overcome loss of voltage-gated sodium channel in Dravet patient neurons (19), and served as proof of principle platforms for using iPSC technologies to study epilepsy. At the same time, they have highlighted potential limitations, such as variability among cell lines, heterogeneity of neuronal populations, and methodology of phenotypic interrogation.
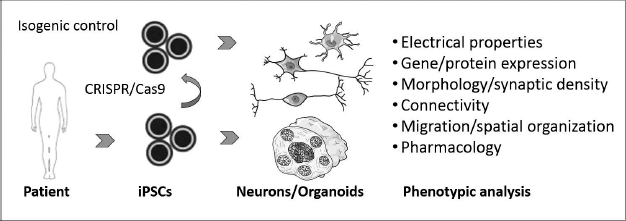
Schematic of iPSC in vitro modeling assays. Isogenic controls are generated using CRISPR/Cas9 gene editing. iPSCs can be differentiated into distinct cellular subtypes such as excitatory neurons, inhibitory neurons, astrocytes, as well as multicellular, 3-dimensional organoids.
Recent advancements in precise gene-editing approaches, including the use of CRISPR/Cas9 (30, 31) and the specific application of this method in pluripotent stem cell lines (32), has enabled researchers to directly test the association of specific genetic variants with cellular phenotypes. This is a critical control in iPSC-based disease modeling experiments, as genetic and potentially epigenetic heterogeneity of distinct patient and control iPSC lines contributes to functional variability of differentiated somatic cells such as neurons, thereby confounding evaluation of phenotyping assays (33). The generation of isogenic stem cell lines through gene editing is the most effective way to overcome this issue and demonstrate causality of a phenotype for a particular genetic variant. Correcting a variant in a patient iPSC line can demonstrate necessity, while introduction of the same variant in a healthy iPSC line can evaluate the sufficiency of the variant for an epileptic phenotype. This approach is particularly relevant in the study of variants of unknown significance (VUS), or others, which exhibit incomplete penetrance. Although gene editing in iPSCs is not a trivial task and requires a significant amount of time, resources, and expertise, it should become obligatory for any convincing demonstration of a phenotype for a genetic type of epilepsy. At the same time, we must keep in mind that diseases with incomplete penetrance indicate the existence of genetic modifiers, which contribute to the phenotype. Thus, cell lines from healthy subjects could also be useful in cases when correcting a mutation does not revert the phenotype to healthy control levels but creates an entirely different phenotype. In cases where the genetic variants remain unknown, it is currently recommended to include comparative data generated from iPSC lines derived from two to three patients, two to three healthy controls, and two to three clones per donor (Guidelines for authors: Cell Stem Cell and Stem Cell Reports journals).
The knowledge acquired from traditional developmental biology studies has allowed for the generation of distinct neural and neuronal subtypes and the continuous improvement of differentiation protocols (for a detailed review on iPSC differentiation and direct conversion methods, see [33, 34]). These protocols are typically based on the utilization of small molecules that activate or inhibit specific molecular pathways, or the exogenous expression of cell type-specific transcription factors and miRNAs that turn on transcriptional cascades and allow for the direct conversion of iPSCs or adult somatic cells. It is important to acknowledge that differentiation protocols often result in relatively heterogeneous cultures. The use of antibiotic selection cassettes (35, 36) is a shrewd approach to increasing the homogeneity of the differentiated neuronal population. A critical feature of modeling epilepsies is interrogating the appropriate disease-relevant cell types. Given the clinical presentation of these patients (seizures, cognitive impairment), as well as the focal source of seizures in pediatric epilepsies that often resides in the cortex (1, 37), a logical assumption is to make and examine cortical excitatory and inhibitory neurons. Immunocytochemistry with neuronal-specific immunoreagents is required at a minimum to define the identity and level of homogeneity of the differentiated neuronal cultures. One aspect that is often overlooked is whether the differentiated neurons selected for modeling express the appropriate ion-channels, ion-channel isoforms, modulators, and relevant milieu of factors important for the function of the protein that is mutated. The advent and relative technical ease of bulk, along with single-cell RNA-sequencing techniques, can allow researchers to accurately define the neuronal identity and diversity at the transcriptional level and is highly recommended as an essential, additional control in disease modeling studies.
The simplest iPSC-based models of neurological disease rely on differentiating patient-specific neurons and studying these in isolation, relative to isogenic or healthy controls. This reductionist approach can address how genetic variants affect specific neuronal subtypes autonomously. Co-culture models with distinct neuronal subtypes, such as excitatory glutamatergic (36) and inhibitory GABAergic interneurons (35), allow for more complex multicellular models. Furthermore, co-culture models with primary mouse or rat glial preparations (38) are a reliable approach to mature neurons in vitro, although this constitutes a source of variability given the heterogeneity of the primary preparations. The recent description of human astrocyte differentiation protocols (39, 40) might enable a reconstitution of an all-human culture model. Neuronal subtype-specific reporters (21, 22) or independent generation and labeling of neurons, followed by subsequent co-culture (35), are appropriate to allow for the differential identification of neurons in co-cultures. The degree to which these co-cultures form reliable networks (as well as the timing required for this to become established in vitro) remains unclear. It is widely assumed, although not experimentally fully established, that the iPSC-derived neurons closely resemble their early embryonic and postnatal in vivo counterparts. This aspect might constitute a technical advantage for modeling the onset and developmental trajectory of EEs, which typically occur in the first few weeks to months of life. Ultimately, the selection of cell type and culture method depends on both the mutant gene of interest as well as on the nature of the biological question at hand.
Highlights
iPSC-based models of epilepsy can be used to study disease in human neurons in the context of each patient's unique genetic background.
Precise gene-editing tools and the generation of isogenic controls allow for directly interrogating the functional relevance of disease-associate variants.
Organoid culture protocols have enabled the development of sophisticated cellular model systems that recapitulate early human neurode-velopment with spatiotemporal resolution.
More Sophisticated Three-Dimensional (3D) Models
Recent developments in 3D differentiating iPSC culture systems, termed organoids, have generated renewed excitement in the field; they allow for modeling human CNS development with faithful recapitulation of cellular organization and formation of intricate neuronal networks (for in-depth reviews on organoids see [41, 42]). Organoids recapitulate the complex spatiotemporal resolution of distinct brain regions and can be utilized to study the elaborate interactions between numerous resident neuronal and glial cell types, and how these might be affected by disease-associated genetic variants. Organoid differentiation protocols rely on the inherent self-organizing capacity of stem cells that are triggered when grown in suspension, with or without the concomitant use of small molecule patterning factors. They can be grown in vitro for extended lengths of time and exhibit aspects of early human neurode-velopment with remarkable accuracy, such as an expanded outer subventricular zone and outer radial glia (which are absent from the mouse cortex [43–45]), as well as a time-sensitive interneuron migration (46). While early cerebral organoids (43) exhibited notable heterogeneity between individual cellular bodies as well as within each body with cells representing different parts of the brain, targeted regional specification has improved homogeneity (45, 47, 48). However, the efficient utility of organoid cultures for modeling diseases, particularly ones with subtle phenotypes, is limited by factors including high variability and prohibitive cost. Nevertheless, the continuous development of improved organoid technologies has been progressing at a rapid pace, and we feel confident that these 3D-culture approaches will soon become widely accessible and standardized. Epilepsy is a complex disease, and although its origins might be neuron autonomous, it ultimately is a disease of brain circuitry dysfunction. Therefore, certain aspects of EE disease manifestations—such as the effects of irregular firing patterns on network formation, neuronal rewiring, as well as interneuron migration and spatiotemporal neuronal organization—might be more effectively teased out in a 3D culture system that mimics early human neurodevelopment.
What Does Epilepsy Look like in a Culture Dish?
The general and potentially simplistic consensus of an in vitro epileptic phenotype is the identification of neuronal hyperexcitability. This can be assayed through numerous approaches including classical whole-cell patch-clamp in individual neurons, multi-electrode arrays (MEA) for population recordings, calcium imaging through Ca2+-sensitive dyes and genetically encoded indicators, or high-throughput optogenetic techniques such as the Optopatch (49). Looking for a phenotype of enhanced firing in neurons (induced or spontaneous) may not necessarily be an accurate recapitulation of epilepsy, as this is not chronically sustainable in a human brain. Manipulation of electrogenic proteins (such as ion channels) may acutely produce hyperexcitability. However, stable mutations in these proteins may alter more than just neuronal excitability and lead to changes in the architecture and connections of the neurons temporally or as they develop. It is difficult to ascertain whether such developmental deficits in infants with EE are due to severe episodic seizure activity or if the seizures are a result of maladaptive neuronal network development. Patient iPSC-based models (2D/3D) can be used to effectively address this question because they allow us to define both electrogenic and nonelectrogenic aspects of epilepsy in a spatiotemporal fashion.
Translation to Therapeutics
Despite decades of research and the availability of multiple antiepileptic drugs (AEDs), the understanding and treatment of epilepsy remains inadequate (37). In approximately 30% of patients, the disease is completely refractory to currently available medication. In all cases, it is hard to predict how a patient will respond to a drug, so clinical management often entails a trial-and-error approach. For those patients diagnosed early in their lives, this can have devastating repercussions, including the extended time between diagnosis and effective treatment and potentially severe effects of seizures and side effects of medication on cognitive development. The nature of the disease-causing variant and broader genetic context of each patient, as well as the inherent response of neurons, could be strong determining factors towards how a patient responds to an AED treatment. If that is the case, then patient-specific iPSC-neuronal models of epilepsy could potentially be used as platforms for optimal drug selection. One way to address the predictive ability of iPSC models would be to perform measurements from neurons derived from carefully selected patients (i.e., those with well-characterized clinical responses to AEDs) and establish whether the effects of drugs on the excitability of neurons in the dish correlate with each patient's pharmacological history. If successful, these models will aid in the development of more effective and targeted therapeutic strategies, allowing clinicians to select the best drug or combination of drugs for each individual patient.
Footnotes
Acknowledgements
We would like to thank Dr. Jennifer Kearny for help with generating ![]() , as well as Drs. Linda Laux, John Milichap, and Al George for thoughtful advice and discussions on modeling epileptic encephalopathy with iPSC technology.
, as well as Drs. Linda Laux, John Milichap, and Al George for thoughtful advice and discussions on modeling epileptic encephalopathy with iPSC technology.
The Kiskinis lab is supported by grants from the Dravet Syndrome Foundation, Les Turner ALS Foundation, Muscular Dystrophy Association, the Northwestern University Clinical and Translational Institute and the National Institute of Neurological Disorders and Stroke/National Institute of Aging of the NIH award R01NS104219. Clinical and Translational Institute (NUCATS).