
Abstract
This review poses the question: Does disruption to cognitive brain networks in epilepsy contribute to the problem of comorbid depression? Initial evidence suggests that the network disease that gives rise to seizures has a predilection for the same cognition-related networks that regulate mood, with comorbidity reflective of more extensive disease. Framing both epilepsy and its psychiatric comorbidities in terms of dysfunction in overlapping (cognitive) networks raises the possibility that depression can be a primary feature of the disease in some cases and facilitates an epilepsy classification system where behavioral features of the disorder are embedded in a neurobiological mechanism.
There is increasing recognition that detecting and treating mood disorders is a priority in the clinical care of individuals with epilepsy (1) given that these patients have a 43% greater likelihood of developing depression relative to healthy control subjects (2). Comorbid depression is associated with elevated morbidity and mortality (3), and affected individuals commonly perceive it as more debilitating than their seizures (4). The etiology of the comorbidity, however, remains unclear. The reframing of epilepsy as a disease of brain networks by the International League Against Epilepsy (ILAE) Commission on Classification and Terminology (5) underscores the aim of this review to examine the neurobiology of depression in epilepsy from the perspective of brain network dysfunction with a particular focus on the role of cognition-related networks.
The Zeitgeist of Brain Networks
In neuroimaging terms, ‘brain networks’ are stationary snapshots of fluctuating neural processing, typically derived by averaging the brain's connectivity or activation across a time series of data (6). Cognitive and affective processes are considered to be emergent properties of these networks (6), with behavioral disorder the product of network dysfunction secondary to neurochemical-, structural-, or perhaps genetic-level abnormalities (6). In a seizure, nodes of a diseased network rapidly hypersynchronize, with epileptic activity in one region entraining the rest of the network (7). In particular, epilepsy is characterized by hypersynchrony of large-scale networks involved in cognition (8) that have also been implicated in the pathogenesis of primary depression.
Abnormal Cognitive Networks Underpin the Symptoms of Depression
Cognitive disturbances have been recognized as a core feature of unipolar depression for millennia and continue to form a component of the formal diagnostic criteria (9). Cognitive features take many forms, including rumination, poor concentration, difficulty identifying emotions, biases for negative stimuli, and vague autobiographic recall (10); indeed, some individuals present with a phenotype of depression dominated by cognitive symptoms (e.g., 11–13). Functional neuroimaging indicates that many symptoms of depression can be attributed to a pathological imbalance between two cognition-related brain networks, the autobiographic memory network (AMN) and the cognitive control network (CCN).
The AMN supports self-referential processing, including episodic recall, daydreaming, and introspection (14). It comprises orbitomesial prefrontal cortex (PFC), rostral anterior cingulate cortex, hippocampus, posterior cingulate, retrosplenial cortices, precuneus, and parietal regions important for mental imagery. A midline subcomponent of it is commonly known as the default-mode network (15). In contrast, the CCN supports working memory, attention, and flexible switching between cognitive sets (16), recruiting dorsolateral PFC and dorsal anterior cingulate cortex, with auxiliary nodes in the mesial temporal lobe and intraparietal sulcus. In the healthy brain, these two networks are anticorrelated; that is, they have an inverse relationship whereby when one network is active, the functioning of the other is suppressed (see Figure 1A). In the case of the AMN-CCN, the AMN activates when an individual is preoccupied by introspection, inhibiting the CCN. Conversely, the AMN is suppressed when the CCN is required to marshal the cognitive resources necessary to perform externally focused tasks (17), suggesting that in cognitively demanding situations, self-focused absorption is replaced by increased alertness to the job at hand.
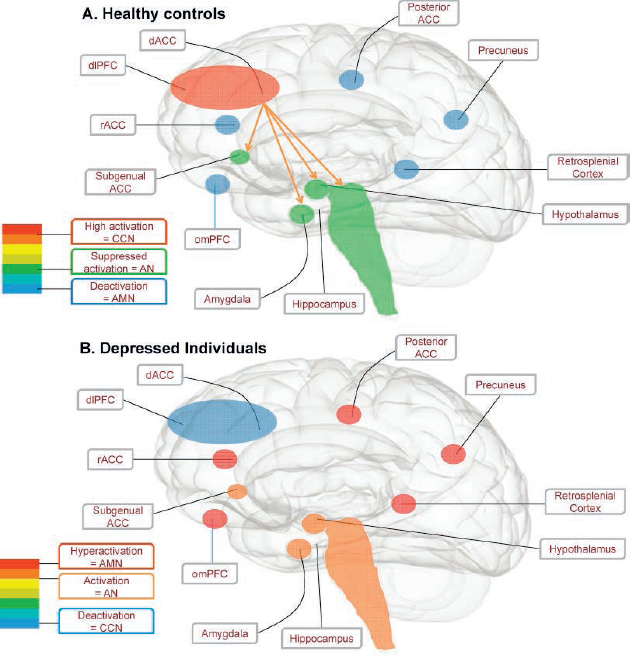
Engagement of the cognitive control network during a non–self-focused task in healthy controls (A) and people with depression (B). (A) In healthy control subjects, activation of the CCN (red) downregulates the AN (green) and is anticorrelated with the AMN (blue). This allows for efficient completion of externally focused tasks. (B) In people with depression, the introspective AMN is pathologically engaged (red), suppressing the activation of the CCN (blue) and leading to uninhibited activation of the AN (orange). In the context of maladaptive core beliefs about the self and the world, this gives rise to symptoms of depression, such as rumination, dysphoria, poor concentration, diminished work performance, self critical information processing. Abbreviations: ACC, anterior cingulate cortex; AMN, autobiographic memory network; AN, affective network; CCN, cognitive control network; dACC, dorsal anterior cingulate cortex; dlPFC, dorsolateral PFC; omPFC, orbitomesial prefrontal cortex; rACC, rostral anterior cingulate cortex. Reprinted from Neuroscience & Biobehavioural Reviews 61, Rayner et al, Cognition-related brain networks underpin the symptoms of unipolar depression: Evidence from a systematic review, 53–65, Copyright (2016), with permission from Elsevier (17).
In depression, AMN-CCN dynamics become skewed. A recent systematic review suggests that compared with healthy controls, individuals with depression have a pathologically overactive and hyperconnected AMN (see Figure 1B) (17). This is linked to features of depression such as brooding, self-blame, and rumination. Simultaneously, abnormally decreased activity is evident throughout the CCN that is linked to poor performance on cognitive control tasks, with heightened CCN activation needed to achieve normal task performance. Symptoms associated with weak task-related CCN engagement include impaired concentration, reduced inhibition of irrelevant negative stimuli, inefficient strategy generation, and slowed processing during effortful tasks.
Disturbed AMN-CCN dynamics in depression have the downstream effect of destabilizing top-down regulation of the affective network (AN) and salience network (SN) (17). The AN overlaps with nodes of both the AMN and CCN (18) and includes bilateral subgenual and pregenual cingulate, ventromesial PFC, and connected regions of the amygdala, entorhinal cortex, hypothalamus, striatum, midbrain, and nucleus accumbens (18–21). These interconnected regions support reward processing and visceral functions such as arousal, appetite, libido, sleep, diurnal variation, and vigilance, disturbances to which underpin vegetative, dysphoric, and anhedonic symptoms of depression (19, 20, 22–24). Highly integrated with both the AN and CCN, the SN comprises the dorsal anterior cingulate cortex (dACC), as well as the left and anterior right insula and the inferior frontal gyri (25). It is thought to be important for detecting stimuli relevant to current goals, initiating cognitive control, and coordinating behavioral responses (25), disturbances to which undermine an individual's capacity to modulate negative emotional responses using cognitive strategies (10, 26). In the healthy brain the activity of the AN and SN are modulated by the CCN (see Figure 1, panel A). People with depression show elevated coactivation between the CCN and AN-SN that is associated with more severe symptoms, heightened monitoring of emotional information, a failure to inhibit irrelevant sad information, and an exaggerated egocentric AMN response to sad information (23, 27; for a review see 17).
Of relevance to epilepsy, framing depressive symptoms as the product of cognitive network disruption provides a potential pathogenic mechanism for depression that is also implicated in the pathogenesis of epilepsy.
Epilepsy: A Disease of Cognitive Networks
Epilepsy can derange the normal organization of cognition-related networks. EEG-fMRI shows that cognitive networks can be coactivated during epileptiform discharges (28), altering their functioning and connectivity over time (29). In particular, epilepsy seems to have a predilection for the networks implicated in depressive symptomatology, the AMN and CCN (30).
Epilepsy and the AMN
Compared with controls, people with focal epilepsy exhibit decreased activation and functional connectivity throughout the AMN (19, 31), suggesting that it is pathologically underengaged. This chronic AMN dysfunction is linked to the habitual deactivation of AMN nodes during epileptiform activity (31, 32). Neuropsychological impairments common in epilepsy that might reasonably stem from AMN dysfunction include poor autobiographic recall, impaired theory of mind, and altered self-related processing (14, 31, 33, 34). The degree of AMN hypoactivation correlates with the severity of cognitive impairment in other neurologic conditions (see 35 for a review), but remains unexplored in epilepsy.
Epilepsy and the CCN
During in-scanner cognitive control tasks, the behavioral performances of people with temporal lobe epilepsy (TLE) are impaired and associated with reduced activation in parietal nodes of the CCN as well as with generalized grey matter loss and diminished white matter integrity within the frontoparietal CCN (36, 37). This may indicate that the structural and functional substrates of the CCN can be disrupted by focal epilepsy, with distal epileptogenic areas potentially accounting for the attentional or working memory dysfunction common to many focal epilepsies (e.g., 38). Abnormal connectivity among the AMN, CCN, and SN has also been reported during both resting-state and task-directed fMRI in TLE (30, 36); however, does dysfunction of cognition-related networks in epilepsy relate to depression in epilepsy more specifically?
Epilepsy + Depression = More Extensive Abnormality of Cognitive Networks?
Neuroimaging Evidence
Preliminary in vivo evidence suggests that depressed patients with epilepsy show neurocognitive network dysfunction beyond what is seen in epilepsy alone. In the most compelling study to date, fluorodeoxyglucose PET in patients with temporal lobe epilepsy revealed frontal lobe disruption specific to patients with depression that mirrors the “hypofrontal” CCN seen in primary depression (39). This has been replicated using both neuroimaging and behavioral methodologies (e.g., 40, 41) and potentially indicates that network nodes implicated in primary depressive disorders may also be relevant to the pathophysiology of depression in epilepsy, with more extensive, anterior network disease symptomatic of mood disturbance.
Tantalizingly, one PET study suggests that cognitive depressive symptoms in people with epilepsy are related to dysfunction in neurocognitive systems distinct from those related to somatic symptoms such as weight loss and health-related complaints. Using a 5-HT1A antagonist in 24 patients with TLE, Lothe et al (42) found that cognitive symptoms correlated with altered serotonergic function in the raphe nuclei and contralateral insula (i.e., SN). In contrast, somatic symptoms correlated with serotonin binding potential in the ipsilateral hippocampus, left mid-cingulate gyrus, and bilateral dorsolateral PFC (i.e., AMN/CCN). Although cognitive symptoms did not map onto AMN/CCN as might be expected, it provides preliminary evidence that depression in epilepsy is linked to disruption in regions known to be pathogenic of depression, with dissociable effects for cognitive versus somatic symptoms.
Behavioral Evidence
Complementing the neuroimaging data, behavioral patterns of cognitive impairment are increasingly used to identify network dysfunction, with cognitive or psychological symptoms providing a marker of the underlying networks that are diseased (43). Cognitive impairments are prevalent in epilepsy, with some degree of network dysfunction detectable in most chronic cases (44). The question for the behavioral method is whether the pervasive cognitive network dysfunction evident in patients with epilepsy is associated with depressive symptoms and, if so, what factors shape the emergence of depression in this subset of patients.
At an individual level, epilepsy can alter select cognitive and affective networks. At a group level, a recent case series reported that nine individuals with frontal lobe epilepsy exhibited significantly poorer cognition and endorsed higher levels of depressive symptomatology relative to 24 healthy controls matched by age, sex, and intelligence quotient (IQ). More in-depth case profiling revealed two distinct patient groups. One was characterized by reduced autobiographic recollection and working memory but a largely euthymic mood, suggesting a cognitive network dysfunction (n=5). A second was characterized by high rates of mood disorder but preserved cognition, indicating an affective network dysfunction (n=4) (23). The dissociation may suggest that discrete neurocognitive or affective networks can be disrupted by the underlying disease in different individuals.
There are other clues that the links between cognitive networks and depression are more nuanced in epilepsy than in psychiatric populations. While some studies show that depressed patients with epilepsy show depression-related cognitive biases, worse cognition, and greater risk of cognitive decline after epilepsy surgery than euthymic patients (e.g., 45–47), others fail to show such a relationship. (e.g., 48). This variability supports the contention that altered cognitive networks are linked to depressive symptoms in epilepsy only in certain subgroups.
Direct support for this observation comes from a study of 85 patients with focal epilepsy compared with 72 healthy control subjects, which found that cognitive network dysfunction is specifically linked to depression in patients with adult-onset epilepsy and a cognitive phenotype of depression. In brief, while the autobiographic memory deficits (AMN dysfunction) of patients with childhood-onset epilepsy were linked to markers of epilepsy chronicity and working memory problems, the autobiographic memory impairments of patients with adult-onset epilepsy were predicted by self-reported depressive symptoms (34). That is, links between cognitive network dysfunction and depression were associated with a clinical feature of the epilepsy, the timing of disease onset. In the same cohort, cognitive network dysfunction was also linked to depression, specifically in patients presenting with a cognitive phenotype of depression (49). Of the 25% of patients with epilepsy who meet criteria for depressive disorder, most (71%) had a cognitive phenotype characterized by cognitive symptoms of depression, such as subjective memory complaints, indecisiveness, poor concentration, and parasuicidal rumination, indicating dysfunction in the AMN and CCN, and was associated with memory impairment on psychometric testing (base rate=17%). The remaining 29% presented with a somatic phenotype characterized by anhedonia and vegetative symptoms like disturbed sleep and appetite, suggesting dysfunction in the AN/SN (base rate=7%). These phenotypes are congruent with those found in other medical and psychiatric populations (11–13) and are consistent with the PET findings detailed earlier (42); thus, it may signify that different presentations of depression in epilepsy indicate dysregulation of different cognitive versus affective networks. These initial insights indicate that depression in epilepsy reflects underlying cognitive network dysfunction in patients with specific neurodevelopmental trajectories and psychiatric profiles.
A Cognitive Network Model of Depression in Epilepsy
Despite the reframing of epilepsy as a disease of brain networks, to date there has been no systems-level model to account for psychiatric comorbidities. Indicators of AMN, CCN, and AN/SN disruption reviewed earlier provide preliminary evidence that the symptoms of depression in epilepsy may result from dysregulation of the same neurocognitive networks implicated in depression more broadly (Figure 1). Although speculative, both the extent of the network disease (as evidenced by fMRI, PET etc) and clinical factors like age at onset may influence the contribution of cognitive networks to depressive symptoms. This raises the possibility that in some cases, cognitive impairment, depressive symptoms, and seizures may arise independently from the same underlying network disease (see Figure 2). This proposition stems from work by Berg (50) as well as Wilson and Baxendale (43), who assert that behavioral features commonly associated with focal epilepsy might be primary manifestations of the disease in some cases, rather than direct effects of overt pathology, seizures, antiepileptic medication, and so on. Secondary interaction effects likely exist in parallel: cognitive and psychiatric impairments are known to lower the seizure threshold (e.g., 42), and there is some literature to suggest that more frequent seizures worsen cognitive integrity (for a review see 44). While the cardinal feature of epilepsy remains the paroxysmal occurrence of seizures, a shared network mechanism may explain the persistent (interictal) cognitive or behavioral disturbances that can be more debilitating than the seizures, as well as account for the observation that cognitive and behavioral symptoms can emerge before the onset of seizures (51, 52). A fundamental task will be to carefully ascertain in vivo how neurocognitive networks change in epilepsy, including determining whether changes to mood-related cognitive networks precede the onset of seizures, indicating that they are an intrinsic feature of the disease.
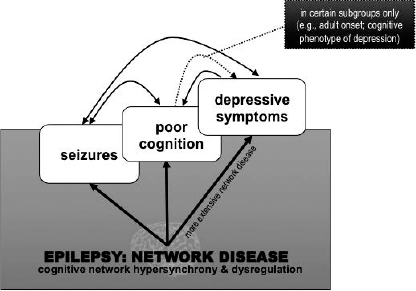
Primary versus secondary effects of network disease on seizures, cognition, and mood.
Conclusion
Systems-level approaches to brain function raise the possibility that depressive symptoms in epilepsy stem from the same diseased networks that propagate seizures, with comorbidity reflective of more extensive disease. Consideration that behavioral disorders may be an emergent property of the network disease heralds a paradigm shift toward a deeper understanding of epilepsy comorbidity based on neurobiology. In due course, this may encourage epilepsy researchers to try to link the cognitive and affective features common across different epilepsy syndromes to intrinsic neurobiological mechanisms, such as genetic abnormalities (53), and calls for more widespread phenotyping of behavioral syndromes in epilepsy in order to identify network abnormalities distinct to different presentations. It is hoped that more precise knowledge of the phenotype-network relationships can lead to the development of medical and psychological treatments targeted to the brain networks implicated in an individual's presentation, ultimately improving patient quality of life.
Highlights
Depression is a common comorbidity of epilepsy
Epilepsy and primary depression are both diseases thought to reflect pathological disruption to cognitive brain networks
Depression in epilepsy linked to more extensive dysfunction of cognitive networks than epilepsy alone
Phenotyping behavioral syndromes in epilepsy may be valuable for narrowing down proximally affected networks
Footnotes
Acknowledgments
I would like to thank Dr Alissandra McIlroy for her helpful discussion and comments on an earlier draft of the manuscript as well as Professors Sarah Wilson and Graeme Jackson for their supervision of the PhD that formed the basis of the current review.
None to declare
None to declare