
Abstract
Absence seizures are common within many different epilepsies and span all the ages. Even though absence seizures were described more than three centuries ago advances associated with its classification, pathophysiology, genetics, treatment, prognosis, and associated co-morbidities continue to be made.
Absence seizures are common in many forms of pediatric and adult epilepsies and are the hallmark seizure type in two epilepsy syndromes—childhood absence epilepsy and juvenile absence epilepsy. The term “absence” was first used to describe seizures in 1705 and was reported as pyknolepsy (“heaped up, closely packed, aggregated attacks”) in the early 20th century (1). Even though more than 300 years have passed since its first description, there continue to be new advances in our understanding of the classification, treatment, pathophysiology, and prognosis of absence seizures and the syndromes they help define.
Clinical Characteristics and Electroencephalography
Typical Absence Seizures
The classical clinical manifestations of typical absence seizures are a transient impairment of consciousness (with abrupt onset and offset) accompanied by one or more other features such as staring, behavioral arrest, eyelid fluttering, or hand/face automatisms (2). The ictal EEG of a typical absence seizure demonstrates generalized spike and wave complexes that are > 2.5 Hz, typically 3–4.5 Hz (3) and lasting ≥ 3 seconds (4, 5). The “3 second” rule is clinically reasonable and provides an objective EEG measure to detect absence seizures when clinical seizures are hard to identify (6). A recent study of 339 seizures in 47 children with absence seizures reported an average ictal duration of 9.4 ± 7 seconds (range 1–44 seconds) (7). Typical absence seizures have a bimodal distribution for age of onset; the first peak at 6–7 years (childhood) with a second peak near 12 years of age (juvenile) (8). The ictal discharges in the juvenile form are slightly less rhythmic and may be of faster frequency (9).
Atypical Absence Seizures
Atypical absence seizures have less abrupt onset and offset, more pronounced changes in tone, variable impairments of consciousness, and tend to last longer than typical absences (10). They are most likely to occur during drowsiness and are not provoked by hyperventilation or photic stimulation (11). Atypical absence seizures have a characteristic interictal EEG pattern with generalized slow (1.5–2.5 Hz) spike and wave complexes that are irregular, asymmetrical, and lower amplitude. The ictal pattern is diffuse, irregular, slow spike and wave complexes that can be associated with irregular diffuse fast activity (5).
Absence with Special Features
The 2010 revised ILAE Report on Terminology and Classification recognized two additional types of absence seizures that have special features: myoclonic absence seizures and eyelid myoclonia with absence (EMA) (12). Seizures described as EMA are characterized as prominent jerking of the eyelids with upward deviation of the eyes, often triggered by eye closure. The ictal EEG pattern for EMA has been described as 3–6 Hz generalized polyspike and wave complexes with occasional paroxysmal bursts in the occipital head regions which can precede the generalized discharges (13).
Syndrome Classification
Childhood Absence Epilepsy (CAE)
CAE is a childhood epilepsy syndrome occurring in 10–17% of all childhood onset epilepsy, making it the most common pediatric epilepsy syndrome (14, 15). Females are more affected than males (16). The ILAE definition of CAE includes very frequent (several to many per day) absences in children of school age (peak manifestation of 6–7 years), and an EEG with bilateral, synchronous, and symmetrical spike-wave discharges at 3 Hz (17). In 2005, an ILAE Task Force for Classification and Terminology added inclusion criteria for age of onset between 4 and 10 years, with a peak between 5 and 7 years (18). It has since become clear that there is a rare subset of patients with onset of absence seizures under the age of 4 years, a proportion of who have glucose transporter type 1 deficiency (19). In addition, the upper age cutoff of 10 years is arbitrary, and there are some who feel that this age limit should not be used to determine which patients to categorize as CAE (3). Instead, it may be reasonable to classify patients with pyknoleptic (very frequent daily) absences, regardless of age, as CAE and there is some indication that onset of a pyknoleptic pattern of absences after age 11 years is unusual (8).
Juvenile Absence Epilepsy (JAE)
Although juvenile absence epilepsy (JAE) also has absences as the main seizure type, the absences of JAE have a less severe impairment of consciousness (even though the duration of electrographic discharges can be longer) and lack a pyknoleptic pattern (i.e., only one or a few absences daily) (20). Most cases begin between 10 and 17 years of age (8). However, at the lower age limit, there is a great deal of overlap with CAE, and it is not clear which criteria—age of onset, pyknoleptic versus non-pyknoleptic events—distinguish between these syndromes for children with onset of absences between 10 to 12 year of age (21). A distinguishing factor from CAE is that generalized tonic clonic seizures (GTCS) are much more common in JAE and have been reported to eventually occur in almost 80% of patients (22).
Jeavons Syndrome
EMA can occur with idiopathic, cryptogenic, or symptomatic epilepsies. The idiopathic form is referred to as Jeavons syndrome, and EMA in this syndrome usually occurs following eyelid closure. Onset is in childhood, and all patients are photosensitive (23). It is unclear whether Jeavons syndrome should be classified as a type of absence epilepsy or as a myoclonic epilepsy, given its prominent eyelid myoclonia.
Pathophysiology
Since the early studies of Jasper and Williams, controversy has ensued regarding whether the structure that produces and controls the spike-wave discharges responsible for absence epilepsy is in the cortex, thalamus, or both (24, 25). Many now accept the unifying hypothesis that the spike-wave discharges of absence seizures are probably produced via reciprocally connected neurons in the thalamus and cortex (26).
Studies of absence seizures using EEG-fMRI have shown activation patterns within the frontal and parietal cortices and thalamus, although the relative amounts of signal increases and decreases have varied (27–29). MEG provides a different approach due to submillisecond time scale temporal resolution (30). A study of children with medically refractory absence epilepsy using MEG showed that there is focal, rather than diffusely generalized, cortical activity prior to the onset of SWDs (31). Similar MEG findings in patients with newly diagnosed and untreated CAE have shown that the area of maximal activity varies depending on frequency bandwidth (Figure 1).
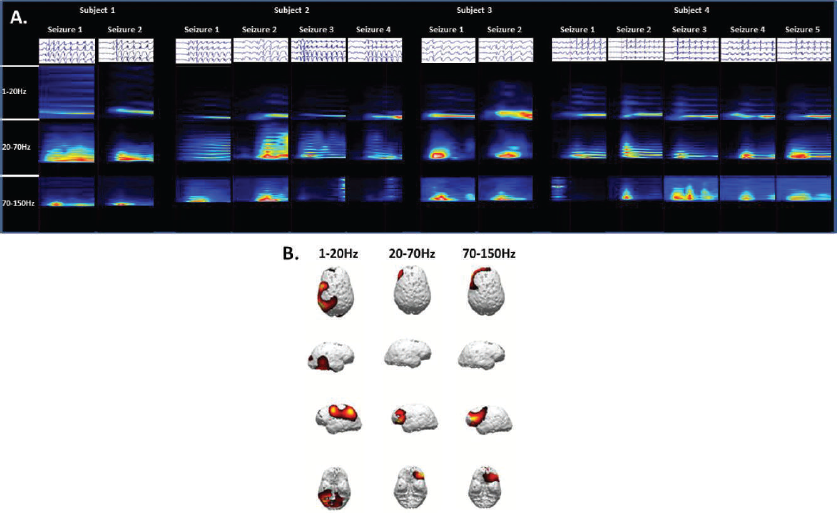
Four children with newly diagnosed and untreated childhood absence epilepsy were recruited and thirteen absence seizures were recorded during the MEG sessions. (A) Time-frequency analysis showed significant spectral power density in the 1–20 Hz, 20–70 Hz, and 70–150 Hz bandwidths. (B) Source localization using a sLORETA algorithm demonstrated consistent localization preferentially in the parietal region for activity at 1–20 Hz and localization preferentially to the lateral frontal region at 20–70 Hz.
Future studies will investigate whether seizure generator location varies among patients, and correlates with either differential comorbidities, differential response to treatment, or differential outcomes.
Genetics
The importance of genetics in the development of absence epilepsies has been recognized for more than 70 years (32). Recent work has underscored the genetic complexity of absence epilepsy. Mutations in genes that code for subunits of GABA receptors, calcium channels and novel non-ion channel proteins have been identified as associated with absence epilepsy but only in small populations of affected patients (33). The unifying mechanistic link between the identified GABA and calcium channel mutations is through disruptive effects on the thalamo-cortical network (33). Given its complexity, a variety of genetic techniques (e.g., whole-exome and whole-genome sequencing, epigenetic studies, copy number variation, etc.) will be needed to better understand the genetics underlying absence epilepsy.
Treatment
Childhood Absence Epilepsy
Prior to 2010, a total of six, short duration, small randomized controlled trials (RCTs) had examined ethosuximide (ESM), valproic acid (VPA), and lamotrigine (LTG) initial monotherapy in children with absence epilepsy. Due to multiple methodological limitations, none of these RCTs met the criteria for either a Class I or II study, providing insufficient evidence to inform clinical practice (34). In 2010, an NIH-funded 446 patient, 32-center double-blind, randomized, comparative controlled clinical trial examined the efficacy and tolerability of ESM, VPA, and LTG (6). At the week 16–20 visit, subjects on ESM (53%) and VPA (58%) had significantly higher freedom from failure rates than LTG (29%, p < 0.001 for both comparisons); subjects on ethosuximide had significantly less attentional dysfunction compared to subjects on valproic acid (33% vs. 49%, p = 0.03). This combination of findings implied that ESM is the optimal initial monotherapy for CAE (6). However, despite being the “winner” at the week 16–20 visit, ESM therapy failed in 47% of subjects (14% due to seizures, 24% due to intolerable side effects, 13% withdrew from study) (6). The recent identification of antiepileptogenic effects of ESM in the WAG/Rij model of absence epilepsy (35, 36) reinforces the need to determine the long-term impact of initial therapy in this large prospectively followed cohort.
Juvenile Absence Epilepsy
No randomized-controlled double-blind trials have been conducted in Juvenile Absence Epilepsy. Separate expert opinion surveys in the US and Europe found VPA and LTG to be the top initial treatment choice for JAE (as they treat both absences and tonic-clonic seizures) (37, 38). Second line (or later) treatments with evidence of modest efficacy for JAE have included ESM, amantadine, and the ketogenic diet (39, 40).
Jeavons Syndrome
Many reports indicate that EMA of idiopathic type or Jeavons syndrome is resistant to pharmacologic treatment (3). Avoidance of seizure precipitants can be important and non-pharmacologic treatments for photosensitive patients, such as wearing special glasses, can be beneficial (41). The most commonly used medications for Jeavons syndrome are VPA, ESM, benzodiazepines (BZDs), levetiracetam (LEV), and phenobarbital (PB).
Outcome and Prognosis
Childhood Absence Epilepsy
Remission rates for CAE, based on epidemiologic cohort studies, range from 21%–74% (42–57). In five prospective cohort studies, the proportion of seizure free subjects were 57%–74% (42, 44–46, 50, 51). Although labeled a “benign” syndrome, the clinical course of CAE is variable and remission rates are far lower than in other classic benign idiopathic epilepsies such as Benign Rolandic Epilepsy (58).
Multiple studies report that GTCs ultimately develop in roughly 40% (range 35%–60%) of children with absence seizures at onset (45, 59–62). GTCs often occur 5 to 10 years after the onset of the absence seizures (45), between 8–15 years old (60, 61, 63). Risk factors include onset of absence seizures after 8 years old, male sex, lack of response to initial therapy and therapy with only an anti-absence drug (8, 62, 64).
Accidental injury is common; 20% of young adults with CAE were reported to suffer an injury during an absence seizure. The risk of accidental injury resulting from an absence seizure is estimated to be 3% per person year (56, 65).
Juvenile Absence Epilepsy
The long-term prognosis for those with JAE is unclear. There is some evidence that patients with only absences have a much greater chance of achieving complete seizure control than those with absences plus GTCS (66). JAE is thought to persist into adulthood at higher rates than CAE. Neurophysiological differences between areas of seizure onset and spread may account for some of the differences in medication responsiveness and long-term prognosis for CAE and JAE.
Jeavons Syndrome
The outcome and prognosis for Jeavons syndrome is poorly understood. There is some evidence that GTCS, either light-induced or spontaneous, will occur in most patients over the long term (67). Jeavons syndrome is thought to be a lifelong disorder, resistant to medical treatment (3).
Comorbidities
Children with CAE have elevated rates of adverse behavioral, psychiatric, language, and cognitive comorbidities (including attentional problems, anxiety, depression, social isolation, and low self-esteem) (68–70). Small series have reported difficulties in the areas of visual attention and visuospatial skills (71, 72), verbal learning and memory (73), and reductions in language abilities (74). The 2010 Childhood Absence Epilepsy study detected overall normal cognition, but 35% of subjects had pre-treatment attentional deficits that did not abate even when seizures were controlled (6).
Conclusion
Although absence seizures are common within many different epilepsies and span all ages, there are still many unanswered questions. Despite many advances during the past decade, there is still much to learn. For example, should criteria for CAE or JAE syndrome classification be based on age at onset, frequency of seizures, or a yet undetermined biomarker? What is the true long-term medication responsiveness and prognosis of CAE and JAE? Many clinical and electrographic features of CAE and JAE are similar, especially at the time of initial diagnosis, but prognosis can vary from complete seizure freedom to medically intractable, lifelong seizures. Are there neurophysiologic differences in the brain networks responsible for generating seizures in these groups of patients? The ongoing Childhood Absence Epilepsy Study aims to address many of these issues for CAE, but the JAE questions remain unanswered. A better understanding of the pathophysiology and course of absence seizures and its syndromes is critical to developing therapies that improve these children's quality of life and potentially the lives of all patients with generalized epilepsies.