
Abstract

A New Mode of Corticothalamic Transmission Revealed in the Gria4−/− Model of Absence Epilepsy.
Paz JT, Bryant AS, Peng K, Fenno L, Yizhar O, Frankel WN, Deisseroth K, Huguenard JR. Nat Neurosci 2011;14:1167–1173.
Corticothalamocortical circuits mediate sensation and generate neural network oscillations associated with slow-wave sleep and various epilepsies. Cortical input to sensory thalamus is thought to mainly evoke feed-forward synaptic inhibition of thalamocortical (TC) cells via reticular thalamic nucleus (nRT) neurons, especially during oscillations. This relies on a stronger synaptic strength in the cortico-nRT pathway than in the cortico-TC pathway, allowing the feed-forward inhibition of TC cells to overcome direct cortico-TC excitation. We found a systemic and specific reduction in strength in GluA4-deficient (Gria4−/−) mice of one excitatory synapse of the rhythmogenic corticothalamocortical system, the cortico-nRT projection, and observed that the oscillations could still be initiated by cortical inputs via the cortico-TC-nRT-TC pathway. These results reveal a previously unknown mode of corticothalamocortical transmission, bypassing direct cortico-nRT excitation, and describe a mechanism for pathological oscillation generation. This mode could be active under other circumstances, representing a previously unknown channel of corticothalamocortical information processing.
Commentary
In The Republic, Plato described a “noble lie” as a myth told by the elite to maintain social harmony. Many of us epileptologists have told our students noble lies when trying to explain how certain unexpected pathogenic mechanisms cause seizures. For example, how does an epilepsy mutation that reduces, rather than enhances, excitatory neurotransmission cause seizures when conventional wisdom dictates that seizures result from disinhibition or hyperexcitation? We rationalize that the affected excitatory receptors must activate critical inhibitory processes. Indeed, deactivated inhibitory interneurons may contribute to seizures in severe myoclonic epilepsy of infancy (1). However, what if the deactivated inhibitory system is also required to sustain the seizures? Could this system, in some circumstances, be deactivated to confer hyperexcitation and, in other circumstances, preserved to allow seizure maintenance? Here, Paz et al. validate such a complex noble lie by discovering that the loss of a glutamate receptor subunit expressed in a critical inhibitory nucleus both deactivated and maintained inhibition depending on its excitatory input, thereby promoting the initiation and continuation of absence seizures.
Decades of study have uncovered an incredibly elegant network responsible for generating absence seizures. As depicted in Figure 1, this seemingly simple network contains three components: the cortex (CT), thalamic reticular nucleus (nRT), and thalamocortical relay neurons (TC) located in the ventrobasal thalamus (VB). Depth electrode recordings in two rat models of absence epilepsy revealed that the seizures begin in the deep layers of the somatosensory CT (2, 3). These deep cortical neurons make two important excitatory connections to TC and nRT neurons: The excitation of the gamma amino butyric acid (GABA)-containing nRT neurons provides a critical feedforward inhibition to the TC neurons (CT → nRT → TC). The TC neurons project to the CT to form cortical EEG discharges and, importantly, back to the nRT to provide TC ↔nRT feedback inhibition needed to sustain oscillations, as reviewed in Beenhakker & Huguenard (4).
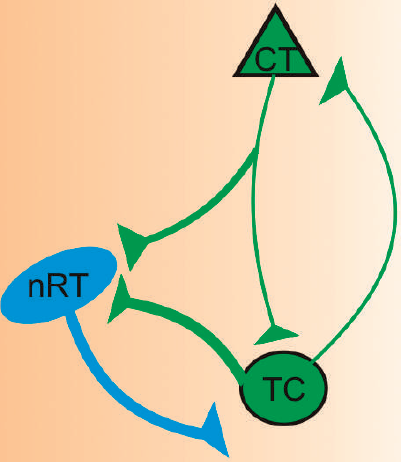
The thalamocortical network involved in absence seizures. The cortex (CT) provides excitatory input to neurons in the thalamic reticular nucleus (nRT), and to thalamocortical (TC) relay neurons in the thalamic ventrobasal nucleus (VB). The nRT provides inhibitory innervation to TC neurons, and TC neurons provide excitatory innervation to nRT as well as to the cortex. Therefore, activation of CT fibers provides feedforward inhibition to TC through nRT, and activation of TC fibers excites the cortex and also provides feedback inhibition through the nRT back to the TC. Ion channels expressed in TC neurons cause them to burst fire upon recovering from hyperpolarization. Therefore, the feedback inhibition from the nRT causes TC neurons to engage in periodic, synchronized burst firing, thereby establishing an oscillation that is transmitted back to the cortex. Reprinted with permission from Paz et al. (10).
What would happen if excitatory transmission were selectively reduced in the nRT? At first, one might construct a noble lie that reducing excitation of the inhibitory nRT would enhance thalamocortical activity and cause absence seizures. However, because of the particular ion channel proteins expressed in TC neurons, coordinated nRT →TC inhibition results in rebound bursting of TC action potentials needed for oscillations and spike-wave discharges (SWD). Therefore, because reduction of nRT excitation would diminish both the CT →nRT →TC feedforward inhibition thought to initiate the TC bursting, as well as the TC ↔nRT feedback inhibition needed to maintain oscillations, it was hypothesized that absence seizures would not occur with nRT deactivation. Of course, nature abhors simple answers. The Gria4 gene—encoding the GluA4 subunit of excitatory 4-amino-3-hydroxy-5-methyl-4isoxazolepropionic acid (AMPA) glutamate receptors—is expressed in substantially greater abundance in the nRT than in other components of the thalamocortical network. Therefore, Gria4 deletion would selectively deactivate the nRT, which would be expected to prevent—not cause—absence seizures. Nevertheless, mice with disrupted or deleted Gria4 possess absence-like SWD and absence epilepsy (5).
To address this apparent contradiction, Paz and colleagues dissected the effects of Gria4 deletion in different components of the thalamocortical network ex vivo in horizontal brain slices that contained the nRT, VB, and the internal capsule that carries the afferent and efferent fibers connecting the thalamus with the cortex. They found that loss of GluA4 expression altered the frequency, amplitude, and time course of current decay of spontaneous excitatory postsynaptic currents (sEPSCs) in nRT but not in TC neurons. This result confirmed that Gria4 deletion did, in fact, selectively reduce excitation in nRT, which would be expected to reduce the propensity for absence seizures. However, extracellular multiunit field recordings with electrodes placed in the nRT and VB demonstrated that electrical stimulation of the internal capsule caused significantly prolonged oscillatory activity in the Gria4−/− thalamus. This result confirmed that Gria4−/− thalami could sustain oscillations despite the reduction of spontaneous nRT excitation, suggesting that TC ↔nRT feedback inhibition remained intact despite a deactivated nRT.
Although measurements of sEPSCs demonstrated reduced spontaneous excitation in nRT, they could not determine if the reduced postsynaptic currents resulted from action potentials originating in CT neurons, TC neurons, or both. The prolonged thalamic oscillations after internal capsule stimulation suggested that nRT was appropriately excited by TC neurons and, thus, TC ↔nRT feedback inhibition was intact. Is it possible that Gria4−/− selectively reduced CT →nRT excitation without reducing TC →nRT excitation? These questions are typically addressed using electrical stimulation to evoke responses from selective, well-defined afferents. However, electrical stimulation is complicated in the thalamocortical network in which thalamocortical and corticothalamic axons are adjacent. For example, electrical stimulation of the internal capsule and recording in TC neurons would stimulate TC neurons by two different mechanisms that would be difficult to distinguish: orthodromic conduction along CT →TC fibers, and antidromic conduction along TC →CT fibers. Paz and colleagues surmounted this limitation using optogenetics. Several weeks before the electrophysiology experiments, they stereotactically injected virus expressing the light-sensitive cation channel, channelrhodopsin-2 (ChR2), in either CT or VB. ChR2 would then be selectively expressed in both the soma and axonal fibers of either CT or TC neurons. Therefore, during the subsequent electrophysiology experiment, axonal fibers that originated from either CT or TC neurons could be selectively activated with photic stimulation. For example, if virus is injected into the cortex, subsequent electrophysiology experiments could provide photic stimulation to the internal capsule and record responses in TC neurons that specifically arose in the cortex (CT →TC axons) without the results confounded by simultaneous stimulation of TC →CT axons, which would then antidromically conduct back to the TC (Figure 2).
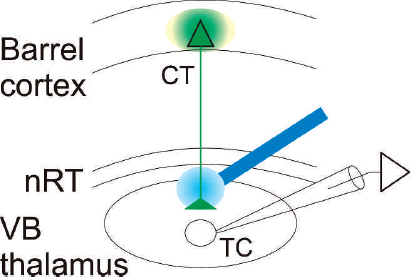
An example of an optogenetic experiment that selectively stimulates cortical fibers and records from TC neurons. Several weeks before the electrophysiological experiment, virus expressing the lightsensitive ion channel, channelrhodopsin-2 (ChR2), is stereotactically injected into in vivo into the area containing the soma of neurons whose fibers are to be stimulated. In this example, virus is injected into the cortex (CT). ChR2 (green) is then selectively expressed in the soma and, importantly, axons of the CT neurons in the internal capsule. Electrophysiology experiments are performed in brain slices containing the thalamic reticular nucleus (nRT), thalamocortical (TC) relay neurons in the thalamic ventrobasal nucleus (VB), and the internal capsule. Photic stimulation (blue) of the internal capsule selectively activates the CT axons but not the adjacent TC axons and, thus, the effect of CT →TC excitation can be accomplished with intracellular recordings in TC neurons. Reprinted with permission from Paz et al. (10).
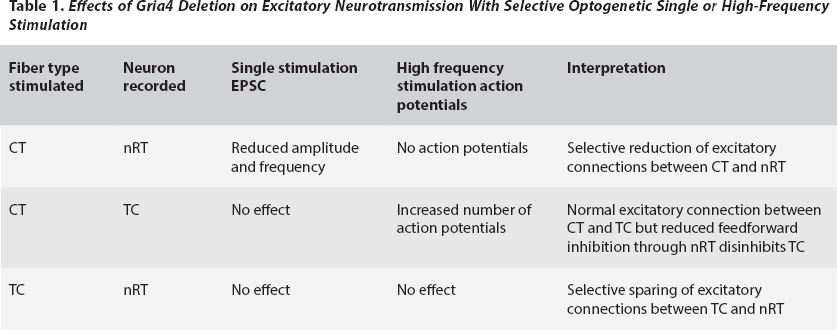
Paz and colleagues applied both single stimulation as well high frequency (70 Hz) photic stimulation to CT and TC axons to evoke EPSCs and action potentials from nRT and TC neurons. The high-frequency stimulation protocol mimicked the high-frequency bursting that occurs in thalamic neurons during spike-wave discharges. These experiments, as seen in Table 1, demonstrated that Gria4 deletion caused three important consequences: 1) attenuated CT →nRT EPSCs with single stimulation and complete failure of nRT action potentials with high frequency CT →nRT stimulation, 2) no effect on CT →TC EPSCs with single stimulation but increased CT →TC action potentials with high frequency stimulation, and 3) no change in TC →nRT EPSCs or action potentials. These results demonstrated that the nRT deactivation identified by the spontaneous EPSCs recordings result from selective loss of CT →nRT excitation without reduction of TC →nRT excitation.
Effects of Gria4 Deletion on Excitatory Neurotransmission With Selective Optogenetic Single or High-Frequency Stimulation
These findings suggested that refinements to the thalamocortical model are needed to explain seizures in this model of absence epilepsy. First, CT →nRT →TC feedforward inhibition is not required to initiate burst firing in thalamocortical neurons. In wild-type mice, strong CT →nRT excitation provides robust inhibition to TC(6). Because recovery from inhibition activates particular TC-expressed ion channels, it was thought that feedforward inhibition initiated bursting. However, in Gria4−/− mice, TC bursting results from direct cortical activation, and not from rebound from CT →nRT →TC feedforward inhibition (Figure 3A). This finding alters our general understanding of TC neuron activation. Second, because Gria4 deletion enhances the activation of TC neurons without affecting TC →nRT excitation, there is stronger TC ↔nRT feedback inhibition (Figure 3B, cycle 1). Third, the bursting TC neurons also activate the cortex, thus providing the SWD seen on EEG. The cortical neurons activated by TC bursts project back to TC to make a mutually excitatory CT ↔TC loop that bypasses the nRT (Figure 3B, cycle 2). It should be noted that this model does not address the timing of CT ↔TC interactions. Is there any substantial latency between the TC →CT bursting and the recurrent CT →TC action potentials in the CT ↔TC loop? If so, could recurrent CT →TC activation take place during a period of TC hyperpolarization and thus interrupt the rebound bursting?
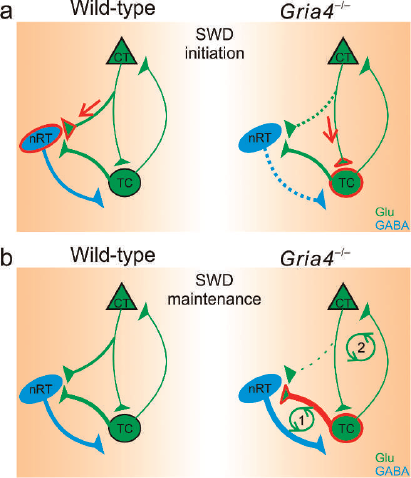
Modified thalamocortical network in the Gria4−/− mouse. A) Spike-wave discharge (SWD) initiation. Wild-type mice (left) have strong feedforward inhibition (red arrow) from the cortex (CT) through the thalamic reticular nucleus (nRT) to the thalamocortical relay neurons (TC). However, because Gria4 deletion (right) selectively reduces CT →nRT excitation (dotted green line), CT axons directly activate TC neurons (red arrow) B) SWD maintenance. Gria4 deletion (right) does not reduce TC →nRT excitation. Therefore, the strongly activated TC neurons receive robust feedback inhibition from the nRT (thick red → thick blue pathway). Upon recovery from the feedback inhibition, the TC neurons engage in synchronized bursting and, thus, establish an oscillation (cycle 1). The oscillation is also projected to the CT (cycle 2). Glu; glutamate CT = cortex, nRT = nucleus reticularis neurons, TC = thalamocortical relay neurons. Reprinted with permission from Paz et al., (10).
The results of this study should prompt us to determine if the selective reduction of nRT feedforward inhibition plays a role in any of the other rodent models of absence epilepsy (2, 3, 7, 8). It is conceivable that even though other pathogenic mechanisms may initiate or sustain seizures in these other animal models, modified feedforward inhibition through the nRT may also contribute to them. Because loss of GluA4 expression selectively reduced excitatory transmission in CT →nRT synapses but not TC →nRT synapses, other glutamate receptor isoforms must be present at the TC →nRT synapses. In animals without the Gria4 deletion, the synaptic expression of the different glutamate receptor isoforms may be differentially regulated. Could a transient downregulation in GluA4 synaptic expression contribute to the formation of seizures in the other models of absence epilepsy?
In addition to determining how the selective loss of nRT feedforward inhibition modulates seizures in other forms of absence epilepsy, it is also important to elucidate how other epileptogenic mechanisms contribute to seizures in Gria4−/− mice, or any other system with selective loss of CT →nRT →TC feedforward inhibition. It has been reported that increased tonic inhibition in TC neurons (9) and cortical hyperexcitability (2, 3) are associated with absence seizures. Do either of these mechanisms play a role in Gria4−/− mice? Cortical hyperexcitability deserves special attention. In the revised thalamocortical model of absence seizures (Figure 3), direct cortical activation of TC neurons initiates the absence seizures and TC →CT projections contribute to seizure maintenance. It is possible that altered cortical physiological processes generate the initial activation that bypasses the CT →nRT →TC feedforward inhibition and causes direct CT →TC activation. Future in vivo and ex vivo cortical recordings will be important to define the role of the cortex in this new model of absence seizures.
Although these efforts may help unify different pathogenic mechanisms of absence seizures into a coherent model, it is certainly possible that different components of the thalamocortical pathway are altered in different animal and human absence epilepsy syndromes. One day, even a single absence epilepsy syndrome may be split into several different categories based upon which element(s) of thalamocortical pathway are disrupted. Possibly, each category may have its own preferential treatment. Of course, until validated or rejected, this conjecture will be just another “myth” we tell our students.