
Abstract
In the 1990s there was intense interest in ionotropic glutamate receptors as therapeutic targets for diverse neurological disorders, including epilepsy. NMDA receptors were thought to play a key role in the generation of seizures, leading to clinical studies of NMDA receptor blocking drugs in epilepsy. Disappointing results dampened enthusiasm for ionotropic glutamate receptors as a therapeutic target. Eventually it became appreciated that another type of ionotropic glutamate receptor, the AMPA receptor, is actually the predominant mediator of excitatory neurotransmission in the central nervous system and moreover that AMPA receptors are critical to the generation and spread of epileptic activity. As drugs became available that selectively target AMPA receptors, it was possible to demonstrate that AMPA receptor antagonists have powerful antiseizure activity in in vitro and in vivo models. A decade later, promising clinical studies with AMPA receptor antagonists, including the potent noncompetitive antagonist perampanel, are once again focusing attention on AMPA receptors as a drug target for epilepsy therapy.
AMPA receptors are ion channels localized to excitatory synapses in the central nervous system that mediate fast (millisecond time scale) excitatory neurotransmission (Fig. 1) (1). In excitatory neurons, action potentials evoke the release of glutamate from presynaptic terminals at synapses onto postsynaptic excitatory and inhibitory neurons. Synapses onto excitatory neurons are predominantly located on dendritic spines whereas excitatory synapses onto inhibitory interneurons are located on the aspiny dendritic shafts typical of these cells (2, 3). In either case, the glutamate released from the excitatory neuron axon terminals diffuses across the synaptic cleft and binds to AMPA receptors in the postsynaptic membrane. The AMPA receptors are large multisubunit protein complexes that span the membrane and have an ion-selective central pore that in the absence of glutamate is closed to ion flow. Binding of glutamate causes the AMPA receptors to gate open, which allows cations to flux across the postsynaptic membrane, resulting in a brief depolarization known as the excitatory postsynaptic potential (EPSP). Although AMPA receptors are permeable to sodium, potassium and in some cases also calcium, at resting potential sodium is the main carrier of the depolarizing current. Summation of EPSPs leads to the firing of action potentials by the postsynaptic neuron, completing the transmission of the synaptic signal. Given their role in fast excitatory signaling in the brain, AMPA receptors are a critical component of all neuronal networks. AMPA receptors are as fundamental to brain function as sodium channels but it is important to appreciate the distinction between the two. AMPA receptors mediate synaptic signaling whereas sodium channels are responsible for a neuron's intrinsic excitability. In line with their distinct functions, sodium channels are voltage-gated and they open in response to membrane depolarization. In contrast, AMPA receptors are largely insensitive to membrane potential; as neurotransmitter-gated channels, their opening is controlled by a chemical signal — glutamate, the universal chemical messenger for fast excitatory neurotransmission.
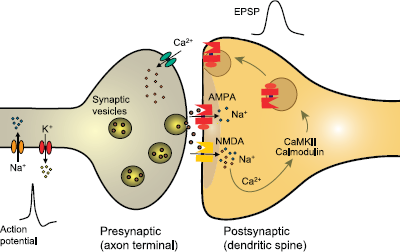
Schematic excitatory synapse. When action potentials mediated by voltage-gated sodium and potassium channels invade the presynaptic terminal, voltage-gated calcium channels are activated. Calcium entering through the channels stimulates glutamate release from synaptic vesicles, which diffuses across the synaptic cleft to activate AMPA receptors, generating the EPSP, a fast membrane depolarization. With strong stimulation, NMDA receptors are activated eliciting various forms of synaptic plasticity. In a simplified view of long-term potentiation, the calcium entering through NMDA receptors binds to calmodulin, which activates calcium- and calmodulin-dependent protein kinase II (CaMKII) that trafficks AMPA receptors into the extrasynaptic plasma membrane from where they become concentrated by lateral diffusion to synaptic sites. The oval abutting the AMPA receptor represents the various auxiliary subunits, including TARPs, cornichon proteins, SynDIG1 and others, that are associated with AMPA receptors and regulate their function and trafficking.
The Architecture of AMPA Receptors
The cloning of the first AMPA receptor subunit in 1989 enabled the structural analysis of AMPA receptors and a detailed characterization of their physiology and pharmacology (4). It is now well established that there are four AMPA receptor subunits designated GluA1–GluA4 (formerly GluR1–GluR4), each encoded by a separate gene (5). The subunits have a modular organization (Fig. 2) (6, 7). There is a large extracellular aminoterminal domain that is involved in receptor assembly, trafficking and modulation; a ligand-binding domain that serves as the recognition site for agonists (including the natural agonist glutamate) and also represents the binding site for competitive antagonists; a transmembrane domain that forms the ion channel (consisting of three membrane-spanning hydrophobic domains and one intramembranous reentrant loop); and a short cytoplasmic carboxy-terminal domain that is involved in targeting the receptor to synapses. The peptide segments connecting the ligand-binding domain to the transmembrane domain transmit conformational changes elicited by agonist binding to the transmembrane ion channel domain, allowing agonist binding to gate the channel to the open state; these segments can be considered the “transducing domain” (8). This region of the channel is critical to binding of noncompetitive antagonists, which prevent channel gating (9).
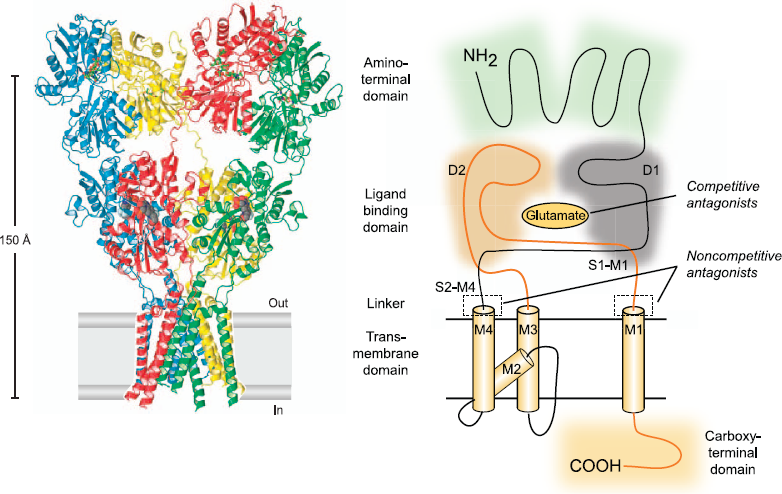
Current model of the protein architecture of the AMPA receptor. Left, view of the broad face of a tetrameric AMPA receptor as determined by X-ray crystallography. Each subunit is in a different color. Adapted from ref. 6 by permission of Macmillan Publishers Ltd., copyright 2009. Right, domain structure of a single AMPA receptor subunit consisting of a bilobed amino-terminal domain, a two-domain ligand-binding core (D1–D2) that serves as the binding site for competitive antagonists, linker segments to which noncompetitive antagonists bind (S1–M1, S2–M4), an ion channel with three membrane-spanning segments (M1–M3, where M3 lines the pore) and a pore loop (M2), and a carboxy-terminal cytoplasmic domain. Agonists, including the natural agonist glutamate, bind to D1 and then the flexible D2 moves towards D1. This movement is thought to pull apart the linkers to the transmembrane channel thus changing the orientation of the transmembrane domains leading to the opening of the ion channel. Binding of noncompetitive antagonists stabilize a configuration of the linkers so that agonist binding fails to induce gating of the channel.
Each subunit consists of approximately 900 amino acids and exhibits 65–75% sequence homology to other subunits. All AMPA receptors are tetrameric combinations of the four subunits. While homomeric receptors are functional, native AMPA receptors are believed to be heteromers. For example, in hippocampal pyramidal cells of mature rats, the most common subunit configurations are GluA1/GluA2 and GluA2/GluA3 (10). GluA2 serves a critical function. Its pre-mRNA undergoes a unique posttranscriptional modification in which coding of a glutamine (Q) in the M2 reentrant loop is changed to that coding for a positively charged arginine (R). When at least one edited GluA2 subunit is present in the tetrameric AMPA receptor, the channel is calcium impermeable; otherwise AMPA receptors are calcium permeable. However, since nearly all GluA2 subunits are edited and the majority of AMPA receptors contain the GluA2 subunit, most AMPA receptors do not flux calcium. However, GluA2-lacking AMPA receptors are common in interneurons and some cortical neurons where their rapid kinetics allows particularly fast synaptic signaling and their calcium permeability mediates novel forms of synaptic plasticity (11).
AMPA receptors are associated with a variety of transmembrane proteins that function as auxiliary subunits, including TARPs (transmembrane AMPA receptor regulatory proteins), such as stargazin; cornichon proteins (CNIH-2, CNIH-3); and SynDIG1 (synapse differentially induced gene 1) (12). The auxiliary subunits regulate channel gating and are involved in subunit folding, assembly, surface expression and the clustering of AMPA receptors at synapses. In addition, the subunits modulate the sensitivity of AMPA receptors to pharmacological agents, including antagonists (13).
Other Ionotropic Glutamate Receptors
AMPA receptors are the most abundant ionotropic glutamate receptors in the mammalian brain. There are, however, two other families, NMDA receptors and kainate receptors, that serve distinct roles. Excitatory synapses generally express both AMPA and NMDA receptors, with AMPA receptors mediating the major component of the synaptic response. NMDA receptors are permeable to calcium as well as sodium and potassium, but they are blocked by magnesium at resting potential. However, repetitive activation of AMPA receptors can cause sufficient depolarization to relieve the magnesium block, enabling a slower NMDA receptor mediated component of the response that is associated with calcium influx into the post-synaptic neuron. This influx triggers various forms of synaptic plasticity, including long-term potentiation (LTP), which results from the activation of calcium-dependent signal transduction cascades that cause trafficking of AMPA receptors into the synapse, thus strengthening synaptic signaling (14). NMDA receptors are the switch that triggers LTP, which is expressed and maintained by the presence of an increased number of active AMPA receptors at the potentiated synapse (15).
Kainate receptors are the least abundant type of ionotropic glutamate receptor. However, they do contribute to excitatory transmission at some synapses (16). In addition, in contrast to the situation for AMPA and NMDA receptors, which are largely but not exclusively postsynaptic, kainate receptors are located presynaptically at both excitatory and inhibitory synapses where they regulate neurotransmitter release (17). Kainate receptors contribute in part to the convulsant actions of kainate, and also to its epileptogenic effects (18). However, kainate also acts on AMPA receptors, albeit at somewhat higher concentrations, and many of its actions are likely to be mediated by AMPA and not kainate receptors. The role of kainate receptors in the pathophysiology of seizures is still incompletely understood. It has been proposed that kainate receptors could contribute to seizure generation in some circumstance (18), but experiments with kainate receptor knockout animals have failed to provide support for such a role in several seizure models (19).
Discovery of Glutamate as a Neurotransmitter
The idea that the amino acid glutamate might serve a special role as an excitatory neurotransmitter began with the observation that glutamate could elicit epileptic phenomena. In the early 1940s, the Japanese physiologist Hayashi observed that clonic convulsions could be induced by injection of glutamate through a fine needle into the gray matter (but not the white matter) of the motor cortex of dogs, and in subsequent experiments a few humans (20). This led Hayashi to propose a model for epilepsy in which epileptic seizures occur when the “quantity of the glutamic acid in human brain surpasses the critical level,” a concept that is still relevant today. Subsequently, Curtis, Phillis and Watkins reported that iontophoretic application of glutamate in the vicinity of neurons in the cat spinal cord caused the neurons to depolarize and fire action potentials (21). However, it was not until two decades later that glutamate was accepted as the major excitatory neurotransmitter in the central nervous system (22). An important step along the way was the identification of a variety of synthetic and naturally occurring neuroexcitatory amino acids related to glutamate. These included N-methyl-D-aspartate [NMDA; synthesized by Watkins (23)], kainic acid (from red alga Digenea simplex) and quisqualic acid (from seeds of Quisqualis plants). A variety of studies with these agents suggested the existence of multiple receptors for the excitatory action of glutamate, which were initially classified as NMDA, kainate and quisqualate after the ligands that selectively activated them. An additional potent excitatory amino acid was a-amino-3-hydroxyl-5-methyl-4-isoxazole-propionic acid (AMPA), which was synthesized by Krogsgaard-Larsen as an analog of ibotenic acid and found to excite neurons through non-NMDA receptors that seemed to be distinct from those activated by kainate (24). However, it was only when selective pharmacological antagonists became available in the 1970s that the current classification of iono-tropic glutamate receptors was solidified. Several antagonists were found to be highly selective for NMDA-induced excitation, which allowed the classification of physiological responses as either sensitive or insensitive to these antagonists, thus enabling the creation of two classes of ionotropic glutamate receptors termed NMDA and non-NMDA receptors. Eventually, antagonists became available that allowed a subclassification of non-NMDA receptor responses into responses mediated by quisqualate or kainate receptors. The current classification therefore recognizes the three families of ionotropic glutamate receptors noted above (NMDA, kainate and quisqualate), except that when quisqualate was found to activate certain metabotropic glutamate receptors—a family of G protein-coupled receptors distinct from ionotropic glutamate receptors that mediate a variety of modulatory actions of glutamate by affecting intracellular signaling mechanisms—the nomenclature was changed so that the term “AMPA receptor” replaced “quisqualate receptor.” Remarkably, when the glutamate receptor subunits were cloned in the late 1980s, three independent but closely structurally related families were defined that corresponded with the classification scheme obtained through pharmacological studies.
NMDA and AMPA Receptor Antagonists
The first group of selective antagonists of NMDA mediated excitation to be defined were structurally related to agonists and included such compounds as D-2-amino-5-phosphonopen-tanoate (AP5) (25) and 3-[(±)-2-carboxypiperazin-4-yl]propyl-l-phosphonate (CPP) (26), which act as competitive antagonists at the glutamate recognition site. Later, dizocilpine (MK-801) and the dissociative anesthetics phencyclidine and ketamine were identified as channel-blocking (uncompetitive) NMDA receptor antagonists (27). However, until the identification by Honoré in the late 1980s of quinoxalinedione competitive non-NMDA antagonists, including CNQX (6-cyano-7-nitroquinox-aline-2,3-dione) and NBQX, it was not possible to selectively block non-NMDA receptors (28, 29). Later, 2,3-benzodiazepines were discovered as a novel class of noncompetitive AMPA receptor antagonists (30). These agents do not interact with the AMPA recognition site. Rather, they block AMPA receptors in a noncompetitive fashion and have been referred to as “negative allosteric modulators.” The prototype GYKI 52466 spawned the development of more potent and selective compounds (31, 32), including talampanel (GYKI 53773; LY 300164), which has been studied in epilepsy clinical trials (Fig. 3) (33). A variety of structurally novel noncompetitive AMPA receptor antagonists have been identified including the highly potent quinazolin-4-one CP-465,022 (34) and the pyridone perampanel (Fig. 3). Since perampanel is currently in late stage clinical development, it is considered in detail.
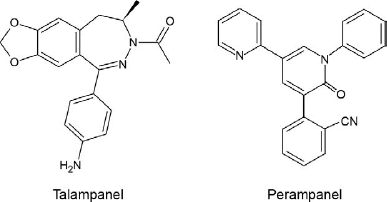
Structures of noncompetitive AMPA receptor antagonists.
Perampanel
Perampanel is a result of a focused discovery program at Eisai research laboratories in London and Ibaraki Prefecture, Japan (35). Structures were explored using high throughput screening assessing inhibition of AMPA-induced cortical neuron death and AMPA-induced calcium influx with the membrane permeant calcium-sensitive dye Fura 2-AM. Structure-activity relationships of compounds based on various core structures were investigated, with the pyridone core eventually being selected as the preferred scaffold. Among a number of potential clinical candidate compounds built on this core, perampanel was selected for further development. Perampanel inhibits AMPA induced calcium-influx into cultured rat cortical neurons in a concentration-dependent fashion with IC50 of 0.093 μM (35). The IC50 value of GYKI 52466 in the same assay is 12.5 μM, highlighting the potency of perampanel. Perampanel caused complex effects on the concentration-response curve for AMPA in this assay so that its precise mode of inhibition has not been definitively defined. However, [3H]perampanel binding was displaced by CP-465,022 and GKYI 52466, indicating that all three noncompetitive antagonists interact at a common site on AMPA receptors. Although perampanel is highly selective for AMPA receptors, high concentrations partially inhibit calcium influx stimulated by the selective kainate receptor agonist (2S,4R)-4-methylglutamic acid (SYM2081), indicating a weak effect on kainate receptors. To date, perampanel has not been found to interact with other molecular targets, including NMDA receptors, at relevant concentrations.
Role of NMDA and AMPA Receptors in Seizure Generation
As the distinction between NMDA and non-NMDA receptors was evolving in the mid-1990s, it became recognized that activation of NMDA receptors induces a pattern of burst firing in neurons reminiscent of the paroxysmal depolarizing shifts (PDS) recorded in the epileptic brain (36). In addition, NMDA receptor antagonists were found to suppress seizure activity in some in vivo and in vitro epilepsy models. Most notably, in in vitro slice preparations, NMDA receptor antagonists inhibited bursting due to direct activation of NMDA receptors (37) and also in the brain slice low magnesium model, in which seizure-like discharges are believed to occur as a result of unblocking of NMDA receptors (38). However, NMDA antagonists alone fail to substantially suppress or eliminate epileptiform activity in most other in vitro seizure models (37). In fact, NMDA receptors antagonists were paradoxically found to enhance the frequency of bursting at the same time that they reduced the burst duration and number of spikes in each discharge (37). In most slice models, it was found necessary to block both NMDA and non-NMDA receptors to fully suppress epileptiform discharges, or in some models blocking non-NMDA receptors alone was sufficient (39–41). In human neocortical tissue removed for the treatment of intractable epilepsy, epileptiform burst discharges were partially sensitive to the NMDA receptor antagonist CPP but were more sensitive to the non-NMDA antagonist CNQX (42). In the case of epileptiform discharges induced by 4-aminopyridine (43) and picrotoxin (39), non-NMDA receptor antagonists alone nearly completely suppressed the discharges (44).
The experience with NMDA receptor antagonists in brain slices presaged the discouraging results of a clinical trial of an NMDA receptor antagonist in which several patients not only did not show improvement but experienced worsening seizures, an increase in epileptiform activity on the EEG, and in one patient the occurrence of complex partial status epilepticus (45). Although NMDA receptor blockade may contribute to the actions of certain antiepileptic drugs (AEDs) that also have other complementary actions (most notably felbamate; 46), it is now apparent that selective NMDA receptor antagonists are unlikely to be useful in epilepsy therapy.
NMDA and AMPA Receptor Antagonists in Animal Seizure Models
Despite the mixed results with NMDA receptor antagonists in brain slice seizure models, NMDA antagonists do confer seizure protection in some animal seizure models, most notably the maximal electroshock test (MES) and reflex seizure models (47). They also have activity in chemoconvulsant models, such as pentylenetetrazol (PTZ)-induced clonic seizures. However, NMDA antagonists are largely inactive against fully kindled seizures and may produce prominent side effects in kindled animals, such as hyperlocomotion and stereotypies (48, 49).
AMPA receptor antagonists have a broader spectrum of anticonvulsant activity (50–53). In fact, unlike NMDA antagonists, they are effective against fully kindled seizures (54, 55). Moreover, they do not produce the side effects seen with NMDA antagonists in kindled animals. The efficacy of AMPA receptor antagonists in kindling models suggests that they might be useful in the treatment of partial seizures. Recent studies also indicate that AMPA receptor antagonists could be effective in the acute treatment of status epilepticus, even when benzodiazepine resistant, and that they may provide a means to avoid the cardiovascular toxicity of benzodiazepines (56, 57). An exception to the broad efficacy of AMPA receptor antagonist is their poor activity in genetic models of absence epilepsy indicating that they will not be of use in the treatment of absence seizures in humans (58, 59).
As is the case with other AMPA receptor antagonists, perampanel is active in the mouse MES test (ED50, 1.6 mg/kg, i.p.) and also effective against audiogenic seizures in mice (ED50, 0.47 mg/kg, i.p.), a reflex seizure model. It was found to be quite potent against PTZ-induced clonic seizures (0.94 mg/kg, p.o.) and to be active in the 6 Hz “psychomotor seizure” test (ED50, 2.1 mg/kg, p.o.) (35). As expected, it is also active in fully amygdala-kindled rats (doses of 5 and 10 mg/kg, p.o., significantly reduced motor seizure duration, afterdischarge duration, and seizure severity) (60); and it had no effect in a rat genetic model of absence epilepsy at doses as high as 10 mg/kg, p.o. (35).
Competitive and noncompetitive AMPA receptor antagonists have similar profiles in animal seizure models. However, in some models, noncompetitive antagonists have improved anticonvulsant activity, which could be due to the inability of high synaptic glutamate levels as occurs during seizures to surmount their blocking action (50). Equivalent protection would require higher doses of a competitive antagonist, but these higher doses would be more likely to induce side effects. In any case, competitive antagonists have not advanced to clinical development because water-soluble forms such as CNQX penetrate the blood-brain barrier poorly whereas lipophilic forms such as NBQX are poorly soluble and precipitate in the kidneys (61).
It has generally been found that AMPA receptor antagonists induce neurological side effects in animal models at anticonvulsant doses, although this varies depending on the seizure model and the test conditions (50, 51, 62). For example, the median toxic dose of perampanel in mice is 1.8 mg/kg, p.o. (35). The toxicity of AMPA receptor antagonists is not unexpected given the critical role that AMPA receptors play in virtually all brain circuits. The lack of a separation between the efficacious and toxic doses naturally leads to the concern that such agents could have poor tolerability in clinical use. However, as discussed below, the available experience from clinical trials indicates that AMPA receptor antagonists are not less well tolerated than many clinically used AEDs, although they do cause sedation, at least transiently, an effect that is also observed in animal models (51). It is noteworthy that AMPA receptor antagonists have not been found to cause PCP-like behavioral effects in animals or psychotomimetic effects in man, as occurs with some NMDA receptor antagonists (63).
Human Studies with AMPA Receptor Antagonists in the Treatment of Epilepsy
Although several AMPA receptor antagonists have undergone clinical trials, only two such agents, talampanel and peram-panel, have been evaluated clinically for seizure prophylaxis. A third compound, the competitive AMPA/kainate receptor antagonist NS1209 has been studied in status epilepticus but the results have not been made available (64). In a small adjunctive trial in patients with refractory partial seizures, orally-administered talampanel appeared to show efficacy (65). However, because of pharmacokinetic interactions (talampanel plasma concentrations are markedly reduced in the presence of enzyme-inducing AEDs), the results were inconclusive. Dizziness and ataxia were the only significant adverse events; importantly, there were no reports of cognitive or psychomotor impairment. However, other studies have noted that talampanel causes drowsiness (66, 67). Additional larger scale trials with talampanel have been completed but since development of the drug has been terminated, it can be concluded that these larger trials were not promising.
Highlights
Glutamate is the principal excitatory chemical neurotransmitter in the brain; AMPA receptors transduce the glutamate signal into a depolarization of the postsynaptic neuron (EPSP).
In addition to mediating the bulk of fast excitatory neurotransmission in the brain, AMPA receptors play a pivotal role in seizure generation and spread.
AMPA receptor antagonists protect broadly against seizures in diverse in vitro and in vivo (animal) models.
Perampanel, a potent noncompetitive AMPA receptor antagonist in late stage clinical development, has demonstrated efficacy and good tolerability in the treatment of refractory partial onset seizures.
AMPA receptors are emerging as a promising new target for epilepsy therapy.
Results with perampanel have been more encouraging and the drug is currently undergoing late stage clinical development. Perampanel exhibits linear pharmacokinetics following oral doses in the range of 0.2 to 6 mg. Maximum plasma concentrations are achieved within 1 h following dosing and the drug has markedly extended kinetics with a mean plasma half-time of 52 to 123 h following a single-dose and 66 to 90 h with multiple-dosing (68). Dose-dependent sedative effects were observed following a single dose in volunteers that were confirmed by demonstrating inhibitory effects on peak saccadic velocity, a sensitive indicator of drug-induced sedation. With chronic dosing, no effects were noted in tests of cognitive performance.
In two phase 2 dose-ranging studies in adults with refractory partial-onset seizures, adjunctive therapy with perampanel at doses of 2 to 12 mg per day was well tolerated (35, 69, 70). Although the studies were not powered to detect statistical significance in efficacy, the proportion of responders—defined as the proportion of patients experiencing reductions in seizure frequency of ≥50%—was larger in perampanel-treated patients than in the placebo group, with a greater proportion of subjects achieving this endpoint in those titrated to 12 mg than in those titrated to 4 mg daily. In a large multinational phase 3 randomized, placebo-controlled adjunctive therapy trial in 712 patients (623 completing the study) with partial seizures receiving 2, 4 or 8 mg of perampanel per day, there was a clear dose-dependent treatment effect in the intent-to-treat population irrespective of whether median seizure frequency or responder rates was used as the endpoint (71). For example, responder rates in the 2, 4 and 8 mg perampanel groups, were 20.9% (not significant), 28.6% (p=0.009) and 34.9% (p<0.001) compared with 17.6% in the placebo group. Dizziness and somnolence were the most common treatment-emergent adverse effects. The number of patients who withdrew due to adverse events was low. There was no greater incidence of serious adverse events in the treatment groups than in the placebo group. Consistent results in terms of efficacy and tolerability were obtained in two additional phase 3 trials (778 patients total) that were similar in design except that perampanel doses of 8 and 12 mg per day were studied.
Conclusions
Disappointing results in clinical trials with NMDA receptor antagonists have raised doubts about the utility of ionotropic glutamate receptors as AED targets. In fact, in retrospect, preclinical studies had provided good reasons to be skeptical of NMDA receptor antagonists. AMPA receptors play a more central role than NMDA receptors in excitatory synaptic transmission, and AMPA receptor antagonists have a broader spectrum of activity than NMDA antagonists in in vitro and in vivo epilepsy models. Moreover, AMPA receptor antagonists do not seem to produce the various troubling neurobehavioral side effects, seen especially in epileptic animals, that are specific to some NMDA receptor antagonists. Recent clinical trials with perampanel, a high potency noncompetitive AMPA receptor antagonist, are encouraging and have renewed interest in ionotropic glutamate receptors as AED targets. Perampanel was discovered by modern methods of medicinal chemistry, in which pharmacodynamic activity at a molecular target is optimized using high throughput in vitro assays (72), an approach that has rarely been successfully applied in AED drug discovery (73). In the years ahead, additional AMPA receptor antagonists will be developed and studied in the clinic. The overall impact of such agents in epilepsy therapy is uncertain but there is cause for optimism given the pivotal role that AMPA receptors play in seizure generation and spread. In addition to their potential utility in seizure prophylaxis, AMPA receptor antagonists represent an intriguing approach for the treatment of status epilepticus. Despite the challenges of clinical trials in status epilepticus, the evaluation of AMPA receptor antagonists in this setting would be worthwhile.
Footnotes
Acknowledgment and Disclosure
The assistance of Eisai in providing published materials on perampanel is gratefully acknowledged; the company did not otherwise contribute to this review. The author has served as a consultant to Eisai for which fees were received.