
Abstract
Primary hyperoxaluria type 1 (PH1) is a rare, inherited, autosomal recessive, metabolic disorder caused by a deficiency of peroxisomal alanine-glyoxylate aminotransferase (AGT). We describe here a case of a 57-year-old man with End Stage Renal Disease, where the late age of presentation of PH T1 due to marked heterogeneity of disease expression caused a delay in diagnosis, and we discuss the causes of the poor outcome typical of this condition
Introduction
Primary hyperoxaluria type 1 (PH1) is a rare, inherited, autosomal recessive, metabolic disorder caused by a deficiency of peroxisomal alanine-glyoxylate aminotransferase (AGT). A hepatic enzyme, AGT catalyses the reaction between L-alanine and glyoxylate to pyruvate and glycine. PH1 is characterized by functional deficiency of hepatic AGT, resulting in overproduction of oxalate and progressive deposition of insoluble calcium oxalate crystals. Early clinical features include urolithiasis and nephrocalcinosis with progressive renal impairment (1). Loss of glomerular filtration rate will lead to impaired oxalate excretion, resulting in systemic oxalosis with oxalate deposition predominantly in bone, skin, retina, myocardium, vessel walls and the central nervous system. We present an atypical case with late diagnosis and discuss management options.
Case report
A 57-year-old man presented to the renal services in October 2007 with End Stage Renal Disease. His past medical history included nephrolithiasis when he was in his 20s; however, he did not remember passing any stones for more than 25 years. His initial presenting complaint was 2-week history of flank pain for which he was prescribed a prolonged course of a nonsteroidal antiinflammatory drug (NSAID) as analgesia. Clinical examination was unremarkable. Laboratory investigations showed serum creatinine 1,334 μmol/L, urea 37.4, potassium 3.8 mmol/L, haemoglobin 5.9 g/dL and corrected calcium 1.62. Immunoglobulin assay revealed low IgM at 0.29 g/L with normal IgA and IgG levels. Serum and urine electrophoresis identified no abnormal bands. Autoantibody screening, including ANA, ANCA, antiGBM and complement levels, was all negative or within normal limits. Ultrasound of the renal tract showed small kidneys bilaterally (right kidney 9 cm, left kidney 8.5 cm) and no evidence of hydronephrosis. The kidneys were grossly echogenic with loss of normal corticomedullary differentiation. There were numerous bilateral renal calculi. A computed tomography (CT) KUB (computed tomography, kidneys, ureters and bladder) confirmed small kidneys, in keeping with advanced chronic kidney disease. It also confirmed the presence of calculi within both kidneys. There were no ureteric calculi. The patient was subsequently commenced on haemodialysis via a tunnelled internal jugular dialysis catheter. The presumed diagnosis was End Stage Renal Disease (ESRD) secondary to nephrolithiasis and NSAID use.
His choice of dialysis was peritoneal dialysis (PD), which was started in November 2007 but had to be abandoned subsequently due to fluid overload, PD peritonitis and subsequent antibiotic-related
He then developed severe bilateral knee pains on exercise with walking distance of around 100 m, which was thought secondary to chronic kidney disease–-mineral bone disorder (CKD-MBD) and high parathyroid hormone 760 ng/L. He underwent parathyroidectomy in 2010; however, his symptoms persisted, and he was referred for specialist orthopaedic opinion. He underwent orthopaedic surgery for spontaneous bilateral patella tendon rupture. At this point, the histology of bone and cartilage samples taken at the time of his knee surgery became available (Figs. 1 and 2). The tibia bone biopsy contained doubly refractile needle-shaped crystals within a fibrous stroma. There was also evidence of foreign body–type giant cell reaction and signs of chronic inflammation. The features were interpreted as showing pseudogout crystal, and he was given analgesics.
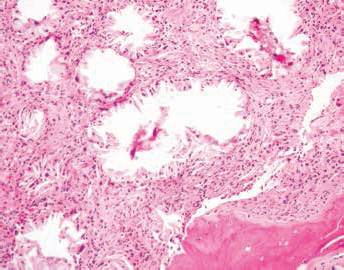
Proximal tibia sample showing bone spicules. There is a bony trabecula at the edge of the biopsy sample.
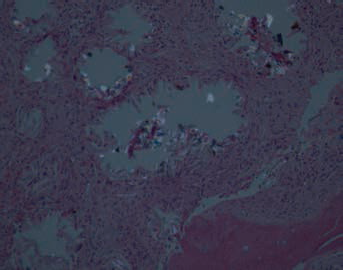
Proximal tibia sample showing needle-shaped crystals under polarised light that are refractile and surrounded by fibrous tissue.
He had been referred to the haematologist team for a review regarding his Erythropoietin Hormone-resistant anaemia. As part of these investigations, he also had a bone marrow aspiration and trephine. The bone marrow examination also showed doubly refractile needle-shaped crystals with giant cell granulomatous reaction (Fig. 3). At this point, oxalosis was for first time considered from the pathology examination of the bone marrow.
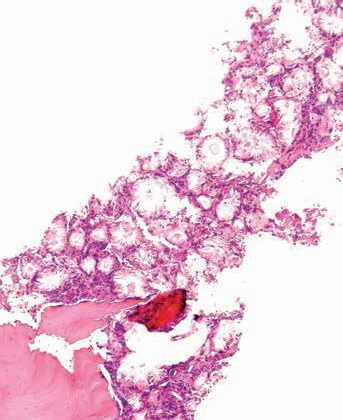
Bone marrow biopsy that showed needle-shaped crystals, but can not polarise refractile material on light microscopy.
The patient underwent genetic analysis which confirmed the diagnosis of PH1. He was then started on high-frequency haemodialysis and was trained to have daily dialysis at his home. He was also started on pyridoxine therapy, and his serum oxalates levels significantly reduced. He was suspended on the renal transplant waiting list and was referred to the liver transplant centre to consider combined liver and kidney transplant. Unfortunately, he died suddenly following an acute cardiac event.
Discussion
PH1 shows considerable phenotypic, enzymatic and genotypic heterogeneity. Some patients have no symptoms at all or suffer only from recurrent urolithiasis. Other patients develop a chronic renal insufficiency with systemic oxalosis. Currently, 3 clinical subgroups are distinguished within PH1. First, there is a rare infantile form with early renal insufficiency (2, 3). Second, there is a rare late-onset form with occasional stone passage in late adulthood and with a good prognosis with respect to renal function. Third, the most common form involves recurrent urolithiasis or nephrocalcinosis and often progressive renal insufficiency. However, the relationship between biochemical and clinical parameters, on the one hand, and outcome on the other, remains unclear.
Even in patients with apparent nephrocalcinosis, conservative treatment with hyperhydration, citrate and pyridoxine can be successful. The heterogenous clinical appearance may explain delay in establishing the diagnosis. Unfortunately, the outcome is poor in most patients. Approximately 50% have been reported to reach ESRD at the age of 25. Patients with ESRD require intensive dialysis sessions to delay systemic oxalate deposition. Renal grafts may fail even after a combined liver–kidney transplantation, due to systemic oxalosis.
In this case, the late age of presentation of PH1 could have been due to marked heterogeneity of disease expression. The patient also had features of bone pain and Erythropoietin Hormone-resistant anaemia which are features of systemic oxalosis (4). The patient was started on daily dialysis. No method of dialysis was shown be to ideal; however, intensive extended daily dialysis, nocturnal dialysis or combination of haemodialysis and PD is recommended to match daily oxalate production (5). The maximal oxalate elimination via conventional haemodialysis and PD is 950 to 1,440 μmol/day, which is significantly lower than daily production of 3,500 to 7,500 μmol/day in patients with PH1 (5). The patient in this case died of acute cardiac event. Cardiac conduction defects may present in PH1, which may result in cardiac arrest (6).
Organ transplantation should be planned prior to systemic oxalosis – i.e., before CKD Stage 5. Ideally simultaneous liver and kidney transplantation is recommended but may be done sequentially depending upon centre.
Footnotes
None.
The authors declare they have no conflicts of interest.