
Abstract
Introduction
Sclerosing angiomatoid nodular transformation (SANT) of the spleen is a rare condition with unknown pathogenesis, which is sometimes associated with hematological disorders. We present the case of renal and splenic AA amyloidosis associated with SANT and essential thrombocythemia – a previously unreported combination of pathologies.
Case description
A 42-year-old Caucasian woman presented with a 6-year history of progressive thrombocytosis, low-grade fever, and elevated C-reactive protein (CRP). An abdominal ultrasound discovered a splenic mass, and a bone marrow biopsy revealed megakaryocytosis with atypical features. Under interferon-α treatment, her fever resolved and her platelet count decreased, but the CRP remained elevated. Within 2.5 years she developed nephrotic syndrome and kidney failure. A kidney biopsy revealed amyloidosis. She was started on hemodialysis and underwent a splenectomy. Splenic pathology revealed SANT and AA amyloidosis of the spleen. Further review of her biopsy specimens confirmed renal AA amyloidosis and myeloproliferative disorder. Polymerase chain reaction studies showed a JAK2 V617F mutation in 1% of nucleated cells. Anti-Epstein-Barr virus (EBV) immunoglobulin G (IgG)-Epstein-Barr nuclear antigen (EBNA) and IgG-viral capsid antigen were >600 U/mL. At the latest follow-up visit 1 year after the splenectomy, she is doing well; her platelet count and CRP are normal.
Conclusions
Our patient has SANT and essential thrombocythemia associated with AA amyloidosis. The high titers of anti-EBV IgG suggest that chronic EBV infection may have been causative for the former 2 conditions. The return of her high CRP level to the normal range after surgical removal of the pseudotumor may suggest an association of AA amyloidosis with SANT.
Keywords
Introduction
Sclerosing angiomatoid nodular transformation (SANT) of the spleen is a rare condition with unknown pathogenesis, which is sometimes associated with hematological disorders. Here, we present the case of renal and splenic AA amyloidosis associated with SANT and essential thrombocythemia – a previously unreported association.
Case description
A 42-year-old Caucasian woman presented in 2007 with asymptomatic progressive thrombocytosis. Four years later, she developed a low-grade fever. Her hemoglobin remained normal, but the platelet count continued to increase, and the C-reactive protein (CRP) was significantly elevated (Tab. I).
Clinical presentation, work-up and interventions
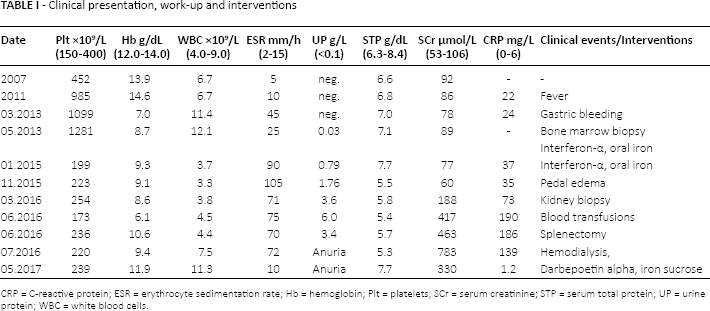
CRP = C-reactive protein; ESR = erythrocyte sedimentation rate; Hb = hemoglobin; Plt = platelets; SCr = serum creatinine; STP = serum total protein; UP = urine protein; WBC = white blood cells.
Two years later, she presented with melena and anemia due to hemorrhagic erosive gastroduodenitis. She received omeprazole and oral iron supplementation. The anemia was partially corrected, but severe thrombocytosis persisted. Ultrasound and computed tomography (CT) showed splenomegaly (87.5 mm2) with a splenic mass that measured 68 mm × 65 mm × 68 mm. A bone marrow biopsy demonstrated increased megakaryocytes with atypical features. She was diagnosed with essential thrombocythemia and started on interferon-α. Her fever resolved, and the platelet count decreased to the normal range; she had mild proteinuria, persistent anemia, and elevated CRP (Tab. I).
After 2.5 years of treatment she presented with pedal edema and increased proteinuria, but with normal serum creatinine and platelet count. The interferon was discontinued. Abdominal ultrasound showed an increase of the size of the splenic mass to 85 mm × 73 mm. Within 4 months she developed nephrotic syndrome, and her serum creatinine increased. A kidney biopsy revealed amyloidosis, but serum and urine immunoelectrophoresis did not demonstrate a monoclonal protein.
Two months later, and 9 years after her first presentation, she was referred to our hospital.
On examination she was conscious, alert, pale, undernourished, with pedal edema, and palpable splenomegaly. She had an otherwise unremarkable physical exam. Her body temperature was 37.1°C, respiratory rate was 17 per minute, pulse was a regular 78 per minute, blood pressure was 110/70 mm Hg, and urine output was 900 mL/24 hours.
She had severe anemia with a normal platelet count, rapidly increasing serum creatinine, and extremely high CRP (Tab. I).
A plain chest x-ray, electrocardiogram, echocardiography, gastroscopy, and colonoscopy were unremarkable. An ultrasound showed splenomegaly with a round, splenic, hypoechoic mass, which was confirmed by abdominal CT.
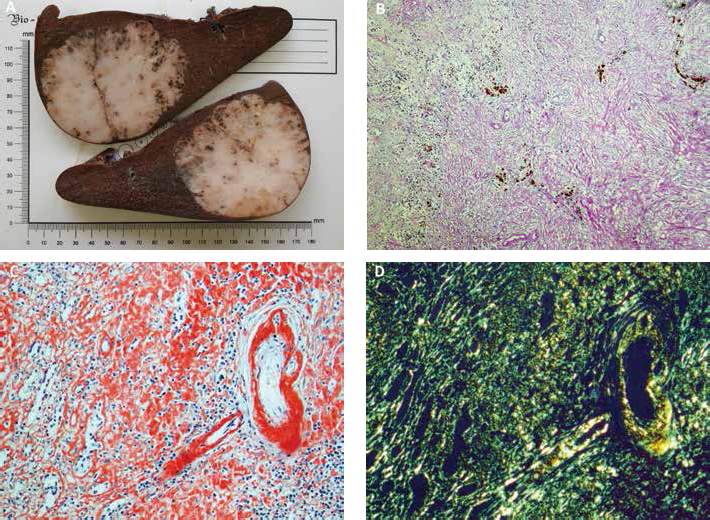
She received blood transfusions; her anemia improved and a splenectomy was performed. The spleen was enlarged, measuring 160 mm × 120 mm × 90 mm, with a dense, round, grey-yellowish mass 90 mm in diameter (Fig. 1A).
(
The postoperative course was complicated by oliguria and a further increase of serum creatinine, and she was started on hemodialysis.
Splenic histology
Light microscopy
There was a solitary vascular splenic mass with slit-like, round, and irregular-shaped vascular spaces surrounded by massive concentric collagen fibers. Vessels were located inside dense fibrous tissue with myofibroblasts, with clusters of plasma cells, lymphocytes, and hemosiderin-laden macrophages (Fig. 1B). There was massive diffuse deposition of amyloid in the stroma and vessel walls of the mass; surrounding splenic tissue was sub-totally involved with amyloid (Fig. 1C D).
Immunochemistry
Immunohistochemistry staining, with an anti-amyloid-A component, CD8, CD31, CD34, CD68, CD163, IgG4, vimentin, desmin, actin, smooth-muscle antigen (SMA) and neuron specific enolase antibodies, demonstrated the AA-type amyloid and features of SANT, with strong CD31 in the endothelium, marked CD163 in the stromal infiltrate, vimentin in the vessels and stromal cells, and actin and SMA in the vessels, with nonspecific false-positive fine granular confluent IgG4 expression in the amyloid deposition areas.
Kidney histology
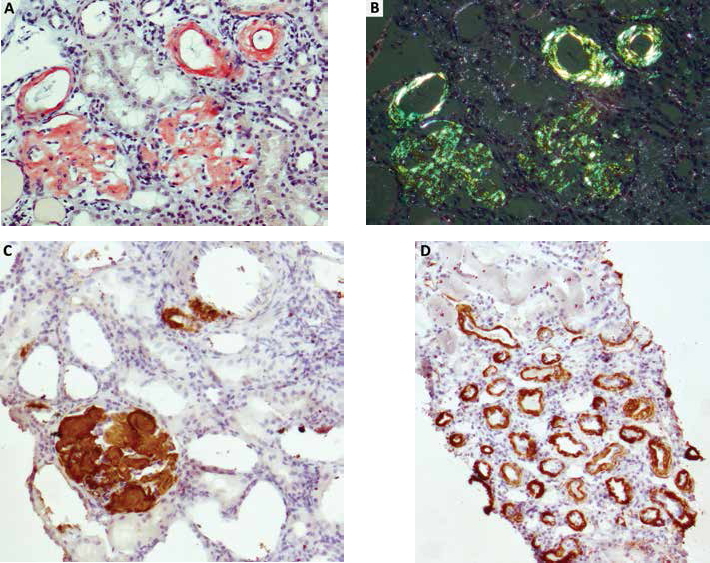
Review of the previous kidney biopsy found 42 glomeruli, 3 of them globally sclerosed, and 7 with segmental sclerosis. Remaining glomeruli were markedly enlarged, with diffuse prominent mesangial, and subendothelial and subepithelial widening due to the deposition of homogenous acellular Congo red positive material, which demonstrated apple-green birefringence under polarized light (Fig. 2A B). There was amyloid deposition along the tubular basement membranes and in the interstitium. Arterioles and small arterial walls were markedly thickened and had narrowed lumens due to amyloid deposition.
(
Immunofluorescence with FITC-conjugated anti-IgA, -IgG, -IgM, -C1q, -C3, -fibrinogen, -λ and -κ light chains antibodies showed nonspecific expression of IgG+++, λ++, and κ++ in the sites of amyloid deposition. Immunohistochemistry with immunoperoxidase staining for amyloid A-protein showed strong diffuse +++ expression of A-amyloid protein (Fig. 2C D).
Bone marrow histology
A review of the previous bone marrow biopsy found increased granulocyte lineage cells. The erythroid precursors were increased with normoblastic erythroblast clusters. There was prominent megakaryocyte proliferation; megakaryocyte lineage elements were in an intratrabecular location – discretely and in small clusters (up to 4 cells). Megakaryocytes were polymorphic with features of atypia: cells of different sizes with hypo- and hypersegmented normochromic nuclei, predominantly large and giant forms with hypersegmented normochromic nuclei, a significant number of anuclear forms, and some megakaryocytes with emperipolesis features. Small lymphoid and mature plasma cells were interstitially dispersed. There were features of a myeloproliferative disorder.
Diagnosis and follow up
Polymerase chain reaction (PCR) showed 1% of cells with a JAK2 gene V617F mutation; there were no mutations of CALR, W515L/K of MPL genes. Anti-Epstein-Barr virus (EBV), immunoglobulin G (IgG)-Epstein-Barr nuclear antigen (EBNA) was >600 U/mL and IgG-viral capsid antigen (VCA) was >750 U/mL, without elevation of anti-EBV IgM-VCA; and PCR for EBV DNA was negative.
Based on these findings, we diagnosed essential thrombocytosis and sclerosing angiomatoid nodular transformation of the spleen with AA amyloidosis with renal and splenic involvement.
At the latest follow-up visit 1 year after the splenectomy, the patient is doing well on hemodialysis in the outpatient unit and has returned to her professional activity. Her body temperature is normal; she gained 3 kilos of dry weight; her anemia is corrected, and her platelet count and CRP are normal.
Conclusions
SANT, described in 2004 (1), is a rare condition affecting usually middle-aged females. It typically manifests a benign clinical course with splenectomy being curative. A total of 134 cases of SANT have been reported in the English language literature thus far. SANT is actually regarded as a specific transformation of splenic red pulp resembling an inflammatory pseudotumor (2-5). Pathology characteristics include multiple angiomatoid nodules forming space-occupying lesions, with 3 immunostaining profiles: capillary (CD31+, CD34+, CD8-), small veins (CD31+, CD34-, CD8-), and sinusoid (CD31+, CD34-, CD8+) (1, 3). SANT was described in association with many conditions, including EBV infection, myelodysplastic syndrome, and idiopathic myelofibrosis (1-5). However, we did not find reports of essential thrombocythemia associated with SANT, nor any descriptions of renal complications or coexisting kidney diseases in patients with SANT.
Myeloproliferative neoplasms (MPN) are infrequently associated with glomerular lesions. Membranous nephropathy, minimal change disease (MCD), focal segmental glomerulosclerosis (FSGS), MPN-related glomerulopathy, and AL (but not AA) amyloidosis have been reported (6-8).
Interferon treatment can be associated with nephrotic syndrome. MCD and collapsing FSGS appearing during interferon treatment have been reported (7). We did not find any prior reports of AA amyloidosis in patients treated with interferon.
Our patient's first presentation was thrombocytosis. Given that 9 years later AA amyloidosis was discovered, she might have had amyloidosis since 2007 with thrombocytosis as a first clinical finding. However, neither clinical features nor work-up favored the diagnosis of any known disease that would cause AA amyloidosis.
In 2007, she might already have SANT with thrombocytosis complicating the chronic inflammatory condition. But this is not consistent with the erythrocyte sedimentation rate, which was normal at that time. Moreover, bone marrow and genetic studies correspond with essential thrombocythemia, even though the PCR test detected only 1% of cells with the JAK2 V617F mutation.
Another scenario not yet described is the association of MPN with renal and spleen AA amyloidosis, the latter mimicking SANT. However, spleen pathology confirmed both the capillary type of SANT and amyloidosis.
Finally, our patient might have had an association of SANT and IgG4-related disease, which is described in the literature (9). But she did not demonstrate any signs or symptoms of IgG4-related disease, and IgG4 expression in the amyloid deposition areas in the spleen was a false-positive result.
Therefore, we consider that she indeed had essential thrombocythemia and SANT, though the causal relationship is not clear. Extremely high titers of anti-EBV IgG antibodies suggest that she had chronic EBV infection, perhaps causative for both SANT and MPN, and was successfully treated with interferon. AA amyloidosis, which complicates a variety of inflammatory conditions, may be causally associated with SANT in our patient via chronic inflammation. Longstanding low-grade fever, high CRP level, and resolution of both findings after surgical removal of the pseudotumor further support a causal association. The mechanism may be similar to Castleman's disease, another nonneoplastic mass with a relatively benign course, accompanied by systemic inflammation and high CRP, for which an association with AA amyloidosis is well described (10).
In conclusion, we present the first reported case of SANT complicated by AA amyloidosis with renal and splenic involvement.
Footnotes
Acknowledgments
We thank doctors Vilen Rameev, Mikhail Tavobilov, Pavel Nikitin, and Alla Kovrigina for their help in the diagnosis and treatment the patient, and our patient for agreeing to the presentation of her data.
Financial support: None.
Conflict of interest: None.