
Abstract
Li-Fraumeni syndrome (LFS) is an autosomal dominant disorder occurring at a young age that predisposes individuals to multiple forms of cancer and to a heterogeneous spectrum of malignancies. We describe the clinical history of a patient who had 5 primary malignant cancers and a familiar history consistent with LFS. We analyzed the genomic DNA of the proband and her relatives by next-generation sequencing (NGS) technology using an enrichment protocol for the simultaneous sequencing of 94 genes involved in hereditary cancers. Genetic analysis of the proband revealed a TP53 germline mutation in exon 5 determining a nucleotide alteration at codon 175 (R175H), a hot spot mutation site related to LFS and a reported pathogenic mutation. The proband daughter's and brother's DNA did not carry the TP53 mutation but they had some rare variants in common with the proband, in addition to other variants with a still unclear role. In conclusion, we identified a TP53 mutation in a patient with multiple primary tumors and a family history characterized by a severe susceptibility to cancer. The genetic analysis by targeted NGS led to the identification of the genetic background and to the exclusion of a cancer risk for the family members. Targeted NGS represents an efficient approach for the identification of mutations in families with a heterogeneous phenotype.
Introduction
Li-Fraumeni syndrome (LFS; OMIM no.151623) is a rare disorder associated with a high risk of developing several types of cancer; in particular, individuals with LFS have an increased risk of developing cancer at a younger age than people who are not affected (1, 2). LFS is often associated with breast cancer at premenopausal age, sarcoma (especially soft tissue and bone sarcomas), brain tumors and adrenal cortical carcinomas (3). Individuals with LFS have a 50%-56% risk of developing multiple primary tumors by the age of 30, which rises to 90%-100% by the age of 60 compared with the general population (4, 5). The estimated risk of developing a second cancer within 30 years of the diagnosis of the first cancer is 57% (6).
Germline mutations in the TP53 tumor suppressor gene (chromosome 17p13; OMIM no.191170) are the molecular basis of LFS and have been identified in 80% of patients with LFS (7). The p53 protein normally controls and regulates cell division and growth through action on the cell cycle and is involved in the repair or destruction of damaged DNA, thus preventing abnormal growth of cells. The TP53 gene consists of 11 exons: 75% of patients affected by LFS have mutations in exons 5-8, which encode the core DNA-binding region of the gene, and 25% show mutations in either exon 4 or 9 (8). Most of the mutations reported are missense mutations (75%), and only a small number of mutations result in premature stop codons and a truncated protein, such as nonsense mutations (9%) and frameshift mutations (6%). Carriers of TP53 mutations have a lifetime risk higher than 90% of developing a malignancy and a 20% risk of developing a tumor before the age of 20 (9).
At the moment, TP53 is the only gene clearly associated with LFS (7), but many other genes have been investigated as possible candidates. Further research is needed to expand the knowledge of the genetic basis of this syndrome in support of the family history and to enable efficient differential diagnosis and accurate treatment.
Case Report
We describe the case of a family with LFS with a peculiar history of tumor development due to the presence of 5 different primary tumors in the same individual and the early development of breast and brain cancers. The study was performed in accordance with the principles of Good Clinical Practice and the ethical standards laid down in the Declaration of Helsinki, and was approved by the Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) Ethics Committee (CE IRST IRCCS-AVR, protocol 2207/2012). The first family member, recruited by our counseling service at the Cancer Prevention Unit of Morgagni-Pierantoni Hospital in Forlì, was a 44-year-old woman (III-1, Fig. 1). She suffered, at the age of 15, of chondrosarcoma. At 35 years old, she was diagnosed with bilateral breast cancer (infiltrating and poorly differentiated ductal carcinoma). Three years later, she developed a superficial bladder cancer (low grade). At the age of 42, she developed a thalamomesencephalic lesion with relatively well-defined margins attributable to cancer of the glial cells that turned out to be an anaplastic astrocytoma grade 3. The patient died at the age of 44.
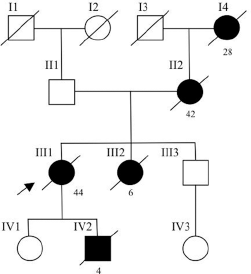
Pedigree of the family with disease-associated TP53 mutation. Circles represent females and squares represent males. Solid symbols represent cancer patients. Symbols with a slash indicate deceased individuals. The arrow points to the proband. I-4 breast cancer (28 y); II-2 bilateral breast cancer (42 y); III-1 chondrosarcoma (15 y), bilateral breast cancer (35 y), bladder cancer (37 y), astrocytoma (42 y); III-2 brain tumor (5 y); IV-1 Down syndrome; IV-2 rhabdomyosarcoma (4 y).
The patient had a daughter (IV-1) suffering from Down syndrome and a son (IV-2) who died at the age of 4 of a rhabdomyosarcoma with relapse. Her sister (III-2) died at 6 years of age because of a brain tumor in the third ventricle (highly differentiated tumor) and her mother (II-2) developed bilateral breast cancer at the age of 42. Her grandmother (I-4) died at 28 years old, probably of breast cancer.
After her informed consent was obtained, the peripheral blood of patient III-1 (proband) was collected. DNA extraction from leukocytes was performed using the QIAamp DNA mini kit (Qiagen). Genetic analysis of the proband was performed using Trusight Cancer (Illumina), an enrichment protocol for the simultaneous sequencing of 94 genes involved in the main hereditary cancer syndromes.
The panel covers a total of 255 kb, on the entire coding regions of 94 genes (AIP, ALK, APC, ATM, BAP1, BLM, BMPR1A, BRCA1, BRCA2, BRIP1, BUB1B, CDC73, CDH1, CDK4, CDKN1C, CDKN2A, CEBPA, CEP57, CHEK2, CYLD, DDB2, DICER1, DIS3L2, EGFR, EPCAM, ERCC2, ERCC3, ERCC4, ERCC5, EXT1, EXT2, EZH2, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, FH, FLCN, GATA2, GPC3, HNF1A, HRAS, KIT, MAX, MEN1, MET, MLH1, MSH2, MSH6, MUTYH, NBN, NF1, NF2, NSD1, PALB2, PHOX2B, PMS1, PMS2, PRF1, PRKAR1A, PTCH1, PTEN, RAD51C, RAD51D, RB1, RECQL4, RET, RHBDF2, RUNX1, SBDS, SDHAF2, SDHB, SDHC, SDHD, SLX4, SMAD4, SMARCB1, STK11, SUFU, TMEM127, TP53, TSC1, TSC2, VHL, WRN, WT1, XPA, XPC). In addition, 100 kb of exon-intron boundaries (50 bp upstream and downstream of each exon) were added to the target regions.
The DNA libraries were prepared starting from 50 ng of genomic DNA and were sequenced on the MiSeq sequencer (Illumina) with MiSeq Reagent Kit v2 (2 × 150 cycles), according to the manufacturer's instructions.
The reads obtained from sequencing were aligned against the human reference genome hg19 with BWA MEM (10). Genome Analysis Toolkit (GATK) version 3.2.2 (11) was used to recalibrate base qualities and realign aligned reads around indels. MarkDuplicates was used to remove duplicate read pairs that arise as artefacts during polymerase chain reaction amplification or sequencing. For variant analysis, GATK UnifiedGenotyper (version 3.5) was used to search for single-nucleotide variants (SNVs) and indels with a minimum fraction of 10% and a minimum base quality score of 15. Genomic and functional annotation of detected variants was done with ANNOVAR (12).
Coverage statistics was performed by the Depth of Coverage utility of GATK. BASH and R custom scripts were used to obtain the list of low coverage (50X) regions per sample. We evaluated the impact of amino acid changes on the 3 patients with predictor tools such as PolyPhen-2 HVAR and SIFT, that use naive Bayes classifiers. Variants were viewed manually on integrative genomics viewer (IGV) to eliminate strand bias and reduce false positive calls. The same next-generation sequencing (NGS) analysis protocol was extended to the brother (III-3) and daughter (IV-1) of the proband.
Confirmatory analysis on the identified TP53 mutation was performed by direct sequencing: the region of TP53 exon 5 was amplified by PCR using specific primers (5’-CTCTTCCTACAGTACTCCCCTGC and 5’-GCCCCAGCTGCTCACCATCGCTA) with Ex Taq DNA polymerase (Takara) and subjected to Sanger sequencing using the BigDye Terminator v3.1 (Life Technologies). The sequences were analyzed by capillary electrophoresis on the 3130 Genetic Analyzer (Life Technologies).
The bioinformatics analysis revealed the presence of 149 exonic variants and 19 splicing variants in the genomic DNA of the proband. To exclude polymorphisms, the identified variants were filtered based on a frequency in the population lower than 0.01 or unknown (Esp6500, 1000genomes and Exac03). The remaining 7 variants were 4 missense mutations in the TP53, BLM, PALB2 and SLX4 genes (c.G524A:p.R175H, c.G3427A:p.E1143K, c.A1001G:p.Y334C and c.C2009A:p.T670N, respectively), 2 synonymous mutations in the ATM and TSC2 genes, and 1 splicing variant in the EGFR gene.
The prediction of the pathogenicity of the 4 missense mutations was concordant for PolyPhen-2 HVAR and SIFT: the mutations in BLM, PALB2 and SLX4 were classified as benign and the mutation in TP53 was classified as deleterious.
The germline mutation in TP53 exon 5, c.G524A:p.R175H (rs28934578), is a hot spot codon commonly mutated in LFS families and was confirmed by a second independent analysis through direct sequencing (Fig. 2).
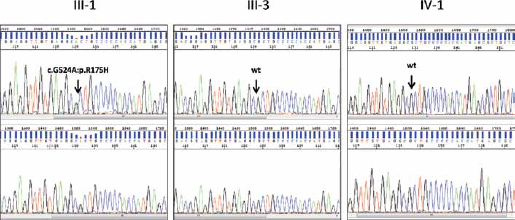
Direct sequencing of TP53 exon 5 showing the heterozygous mutation c.G524A:p.R175H in the proband III-1 (forward strand in the upper electropherogram and reverse strand in the lower electropherogram) compared to the wild-type sequence in her brother (III-3) and daughter (IV-1).
Even though the proband's brother (III-3) and the proband's daughter (IV-1) who suffers from Down syndrome were healthy at the time of the study, in order to assess their risk, they underwent the same NGS genetic testing.
Applying a frequency filter of the identified variants (<0.01 or NA), the cases III-3 and IV-1 were both found to be carriers of the mutations in ATM and BLM identified in the proband. The individual III-3 was also a carrier of the missense mutation in PALB2, while the individual IV-1 was a carrier of the splicing mutation in EGFR. In addition, case III-3 had a missense mutation in the FANCM gene (c.A974T:p.D325V) and 2 synonymous mutations in BRCA2 and ERCC5 (c.C741T:p.I247I and c.T1983C:p.I661I, respectively). Case IV-1 had 4 missense mutations in BRCA1, BRCA2, RAD51C and ERCC4 (c.A4039G:p.R1347G, c.G3581A:p.G1194D, c.G376A:p.A126T and c.C1135T:p.P379S, respectively), 2 synonymous mutations in MSH2 and FANCD2 (c.G2094A:p.E698E and c.G2181A:p.P727P, respectively), and a splicing mutation in EZH2 (c.247-9insT). Table I shows the genetic variants identified in each member of the family.
Genetic variants identified in each member of the family
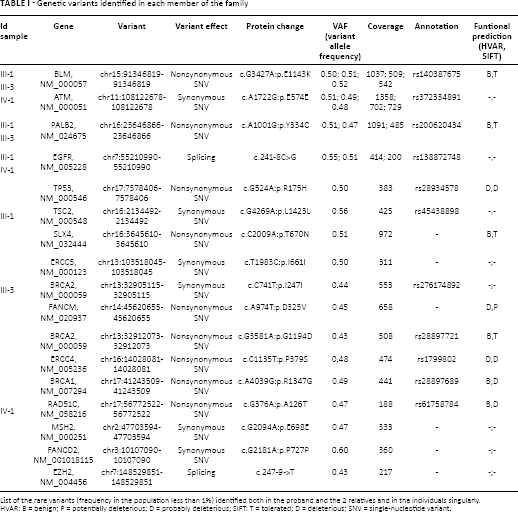
List of the rare variants (frequency in the population less than 1%) identified both in the proband and the 2 relatives and in the individuals singularly.
HVAR: B = benign; P = potentially deleterious; D = probably deleterious; SIFT: T = tolerated; D = deleterious; SNV = single-nucleotide variant.
Prediction of the effect of the missense variants identified in the proband's relatives was concordant for PolyPhen-2 HVAR and SIFT only for the ERCC4 and BRCA2 mutations (classified as deleterious and benign, respectively). It was not concordant for the FANCM mutation (classified as deleterious by SIFT and possibly deleterious by PolyPhen), BRCA1 and RAD51C mutations (classified as deleterious by SIFT and benign by PolyPhen).
A second analysis in individuals III-3 and IV-1 by direct sequencing on TP53 exon 5 confirmed that they were not carriers of the R175H mutation in TP53 (Fig. 2).
Discussion
We have described the family history and genetic characterization of a 44-year-old female patient who developed 5 primary malignant tumors during her lifetime. The proband showed the germline mutation c.G524A:p.R175H in the TP53 gene, which lies in a hot spot codon commonly mutated in sporadic cancers. The protein with the R175H mutation failed to fold properly, resulting in a significant loss of transcriptional and tumor suppressive activity (13). In the literature, the reported mutation is indicated as responsible for the clinical history of aggressive tumors in LFS families (14) and is frequently associated with breast cancer, brain cancer and soft tissue cancer. All these findings are consistent with the type of cancers diagnosed in the family described. The development of such a large number of primary tumors, the association with the mutation in TP53, and the family history make this a model case of LFS, and therefore interesting to be thoroughly genetically characterized.
In order to clarify the causes of the genetic predisposition to multiple tumors in the proband and the segregation pathway in relatives, we performed a comprehensive investigation of the family genetic background by targeted NGS, expanding the analysis to 94 genes involved in multiple hereditary cancer syndromes.
Effect prediction of the mutations found in the proband in addition to the TP53 mutation was possible only for the BLM, PALB2 and SLX4 missense variants, and they were classified as benign. The mutations in BLM, PALB2, ATM and EGFR were also present in one or both of the proband's relatives who were in good health, and therefore do not seem to segregate with the syndrome.
The analysis of TP53 gene mutation in other family members allowed to exclude a cancer risk in the proband's healthy brother (III-3) and consequently in his daughter (IV-3). Moreover, the Down syndrome case (IV-1) was not associated with the TP53 mutation identified. In addition, in cases III-3 and IV-1 we found 10 rare mutations that were not present in the proband and whose role in the cancer risk is unclear. The prediction of the effect of these alterations by bioinformatics tools is not always concordant and the lack of data on other members of the family prevents us from drawing any further conclusions. Unfortunately, it was not possible to analyze the sister (III-2) and the son (IV-2) of the proband for TP53 gene mutation but, given their personal history of cancer, they were also likely to be carriers of the mutation.
Analysis of the genetic background of the proband provided the identification of the causal mutation of the syndrome but also other genetic alterations, including mutations in BLM, PALB2, SLX4 and ATM. Many studies have reported an increased risk of breast cancer associated with mutations of these genes (15-16-17-18), which is consistent with the family history described here, where 3 individuals were affected by breast cancer (III-1, II-2 and I-4).
Conclusion
Although it was not possible to recover DNA from all family members, the genetic analysis of a broad spectrum of genes involved in hereditary cancers, combined with the family history, allowed to outline a more accurate diagnosis and to assess the risk of specific cancers. The analysis of Multi-gene panels is an important tool to gain insight into the mechanisms that lead to a high susceptibility to certain tumors and the interactions between causative mutations. Moreover, background mutations increase the knowledge of the connections between the genotype and the phenotype of the family.
Footnotes
Abbreviations
Financial support: No grants or funding have been received for this study.
Conflict of interest: None of the authors has any financial interest related to this study to disclose.