
Abstract
Germ cell tumors (GCTs) generally express wild-type p53 protein. Rare p53 mutations may be associated with cisplatin resistance. There is growing interest in the role of cyclins as targets for GCTs. Cyclin B1 is involved in G2/M transition and its overexpression has been reported in tumors carrying nonfunctional p53. Conversely, cyclin B1-specific small interfering RNAs have been shown to dramatically reduce tumor proliferation. We investigated whether a subset of chemotherapy-resistant GCTs overexpressed cyclin B1 as a result of nonfunctional p53, as this would make cyclin B1 a potential therapeutic target. Our data showed that GCTs consistently overexpressed cyclin B1 independently of their responsiveness to chemotherapy or the presence of p53 mutations. Cyclin B1 was overexpressed by GCT cell lines carrying functional p53. Cyclin B1-specific small interfering RNAs only slightly reduced the proliferation of JAR and JEG-3 placental choriocarcinoma cells. Further research into targeting cyclin B1 could provide a novel intervention for GCTs.
Keywords
Introduction
Germ cell tumors (GCTs) are a heterogeneous entity composed of different histological subtypes: seminoma, embryonal carcinoma, choriocarcinoma, yolk sac tumor, and teratoma (1). Compared with most solid tumors, GCTs are highly sensitive to cisplatin-based chemotherapy, resulting in cure in almost 80% of patients with advanced disease (1). The low incidence (<7%) of p53 mutations coupled with a high basal expression of functional p53 have long been considered factors contributing to GCT sensitivity to DNA damage by chemotherapeutic agents (2-4). This belief was recently challenged by immunohistochemical analysis of platinum-responsive and platinum-refractory disease, the results of which suggest that the p53 level alone does not account for the cisplatin sensitivity of GCTs (2-4). Conversely, mutations in the p53 gene are likely to account for chemoresistance only in a proportion of cases (2-4).
Cyclins are a family of molecules that control the cell cycle by associating with and activating cyclin-dependent kinases (CDKs). Deregulated expression of cyclins and/or CDKs may disrupt cell growth and lead to a malignant phenotype (5). Cyclin B1 is involved in the transition from G2 to M phase of the cell cycle (6). Wild-type p53 expression prevents G2/M transition in mouse and human cell lines by lowering the intracellular levels of cyclin B1 (7, 8). In human tumors, an inverse correlation has been found between normal p53 function and cyclin B1 overexpression (9). Cyclin B1 depletion by specific small interfering RNAs (siRNAs) has been shown to inhibit the proliferation of human tumor cells (10, 11). We hypothesized that a fraction of GCTs may overexpress cyclin B1 as a result of nonfunctional p53, thus displaying its potential as a therapeutic target. To confirm the hypothesis, we evaluated surgical GCT specimens and cell lines for cyclin B1 and p53 expression, and subsequently used siRNAs targeting cyclin B1 to reduce its expression and assess the impact of this treatment on cell proliferation.
Materials and Methods
Tumor Sections
Archival paraffin-embedded sections of GCT encompassing different histological types according to the WHO 2004 classification were selected for immunohistochemical analysis. Sections from other tumor types and normal tissues were used as controls. Table I summarizes the patient characteristics.
Patient characteristics
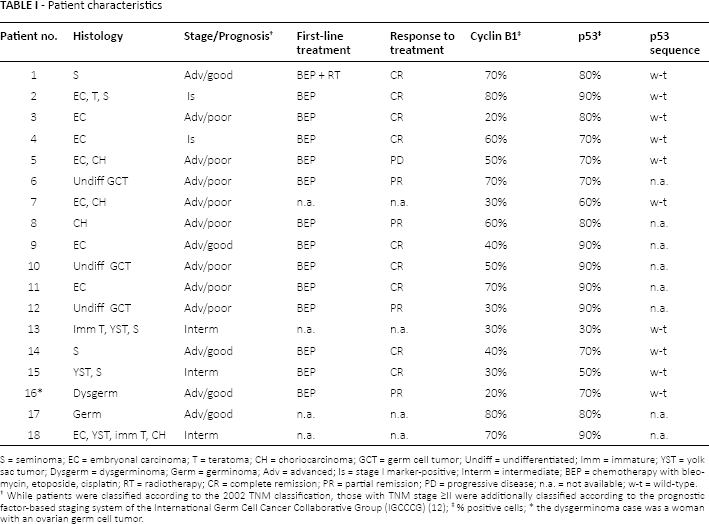
S = seminoma; EC = embryonal carcinoma; T = teratoma; CH = choriocarcinoma; GCT = germ cell tumor; Undiff = undifferentiated; Imm = immature; YST = yolk sac tumor; Dysgerm = dysgerminoma; Germ = germinoma; Adv = advanced; Is = stage I marker-positive; Interm = intermediate; BEP = chemotherapy with bleomycin, etoposide, cisplatin; RT = radiotherapy; CR = complete remission; PR = partial remission; PD = progressive disease; n.a. = not available; w-t = wild-type.
While patients were classified according to the 2002 TNM classification, those with TNM stage ≥II were additionally classified according to the prognostic factor-based staging system of the International Germ Cell Cancer Collaborative Group (IGCCCG) (12);
% positive cells;
the dysgerminoma case was a woman with an ovarian germ cell tumor.
Cell Lines
The human placental choriocarcinoma cell lines JAR and JEG-3, the human breast carcinoma cell line MCF-7, and the human cervical carcinoma cell line HeLa were purchased from ATCC (Manassas, VA, USA). The human testicular embryonal carcinoma cell lines 2102EP (cisplatin-sensitive) and 1411HP (cisplatin-resistant) were kindly provided by Dr. Thomas Mueller (Halle, Germany). IC90 determination (i.e., the cisplatin concentration inhibiting cell growth by 90%) revealed that the 1411HP cell line had 3.3-fold higher cisplatin resistance than the cisplatin-sensitive cell line 2102EP (13). Cells were cultured in Iscove's medium supplemented with 10% fetal bovine serum, glutamine, antibiotics and HEPES.
Immunohistochemical Staining of Cyclin B1 and p53 in Tumor Sections and Tumor Cell Lines
Tumor samples were sectioned (3-5 µm), dewaxed in xylene, and rehydrated through a series of graded alcohols according to standard histological procedure. Antigen retrieval was performed using a pressure cooker and citrate buffer solution (pH = 6), after which tissue sections were submitted to the immunohistochemical procedure. Cell lines were cultured for 48 hours on cell culture slides and then fixed in 95% alcohol for 20 minutes at room temperature. Tissue sections and cell culture slides were then stained with anti-cyclin B1 monoclonal antibody (mAb, Clone GSN-1, BD Pharmingen, San Diego, CA, USA) at 1:300 dilution and/or anti-p53 mAb (Clone DO-7, NeoMarkers, Fremont, CA, USA) at 1:100 dilution. Immunostaining was performed on an automated immunostainer (Autostainer Plus, Dako, Fort Collins, CO, USA) using the ChemMate alkaline phosphatase/red detection kit (Dako, Glostrup, Denmark). Cytoplasmic staining for cyclin B1 was considered positive. A cutoff of 10% was chosen on the basis of normal tissue staining. Red nuclear staining was considered positive for p53 protein. For each case, the percentage of positive cells was evaluated by examining different areas (6-8) of the tissue section and counting at least 800-1,000 tumor cells. Palatin tonsils were used as positive control while negative control tests were performed by omitting the primary antibody.
Western immunoblotting of p53 and cyclin B1
Total cell lysates were separated on a 10% SDS (sodium dodecyl sulfate)-polyacrylamide gel and transferred to nitrocellulose. The filters were blocked in phosphate-buffered saline (PBS) with 5% skim milk and then incubated overnight with primary antibodies specific for p53 (NeoMarkers) and cyclin B1 (BD Pharmingen). The filters were incubated with secondary peroxidase-linked whole antibodies (Amersham Biosciences Europe GmbH, Freiburg, Germany). Bound antibodies were detected using the enhanced chemiluminescence Western blotting detection system (Amersham Biosciences). Results were quantified by densitometric analysis using the ImageQuant TL software (Molecular Dynamics, Sunnyvale, CA, USA).
DNA isolation and sequencing of p53 exons 5 to 9
Genomic DNA was obtained by proteinase K digestion, phenol-chloroform extraction and ethanol precipitation from 10 formalin-fixed tumor samples for which sufficient material was available. p53 mutational status was assessed by direct sequencing of genomic DNA spanning exons 5-9, where >95% of human mutations are detected. PCR reactions were carried out using 50 ng of DNA, 0.8 U of Taq DNA polymerase with proofreading activity (TaKaRa LA Taq, Cambrex, Milan, Italy), 0.4 µM of each forward and reverse primer, 1X LA Taq buffer, and 1.5 mM MgCl2 in a total volume of 20 µL.
PCR conditions were as follows: 2 minutes at 94°C followed by 20 cycles of 30 seconds at 94°C, 45 seconds at 63°C (the annealing temperature was reduced by 0.5°C every 3 cycles), and 1 minute at 72°C. The reaction was continued by 30 cycles of 30 seconds at 94°C, 45 seconds at 60°C, and 1 minute at 72°C.
Aliquots of the amplification reactions were loaded onto 1.5% agarose gels to verify the presence of the expected band and the remaining sample was purified using Qiagen purification columns. Sequencing was performed with an ABI 3730 DNA sequencer. Both strands were sequenced for each exon and for each patient.
The following primers were used:
forward exons 5 and 6: 5’ TGTTCACTTGTGCCCTGACT
reverse exons 5 and 6: 5’ TTAACCCCTCCTCCCAGAGA
forward exon 7: 5’ AGGCGCACTGGCCTCATCTT
reverse exon 7: 5’ TGTGCAGGGTGGCAAGTGGC
forward exons 8 and 9: 5’ TTGGGAGTAGATGGAGCCT
reverse exons 8 and 9: 5’ AGTGTTAGACTGGAAACTTT
siRNAs, in vitro transfection, proliferation assay and Western blot analysis
Two siRNAs targeting cyclin B1 (National Center for Biotechnology Information-NCBI accession number: NM 031966) were synthesized by Dharmacon Research, Inc. (Lafayette, CO, USA): siRNA1 corresponds to positions 340-360 of the cyclin B1 open reading frame, and siRNA3 to positions 776-796. Control siRNAs targeting green fluorescent protein (GFP) were purchased from Dharmacon. All siRNAs were 21 nucleotides in length and contained symmetric 3’ overhangs of 2 deoxythymidines. siRNA transfection was performed as previously described (9). In brief, 24 hours prior to transfection, cells were seeded without antibiotics to a density of 50%-60%. Cells were transfected with 10 nM of the siRNAs using the transfection reagent oligofectamine according to the manufacturer's instructions (Invitrogen). Control cells were treated with oligofectamine alone. Cells were harvested 48 hours after siRNA treatment for Western blot analysis and for the determination of cell numbers using a hemacytometer. To determine the transfection efficiency, cells were treated with 20 nM of each FITC-labeled siRNA for 6 hours, after which positive cells were evaluated with a Becton Dickinson FACScan flow cytometer (BD Biosciences, Heidelberg, Germany).
Results
Cyclin B1 is overexpressed in the cytoplasm of human GCTs and cell lines
Sections of paraffin-embedded samples of 18 GCT patients stained with anti-cyclin B1 monoclonal antibody. Examples of cyclin B1 overexpression are shown in Figure 1A-D. Tumor cells in all specimens expressed variable but easily detectable levels of cyclin B1, whereas cyclin B1 was virtually undetectable in the surrounding normal cells. These findings are in agreement with those from previous articles in which cyclin B1 protein was not detected in Leydig, Sertoli or maturing germ cells in the center of seminiferous tubules but showed heterogeneous expression in proliferating germ cell compartments (14). The percentage of positive cells varied between 20% and 80% (mean 50%) (Tab. I).
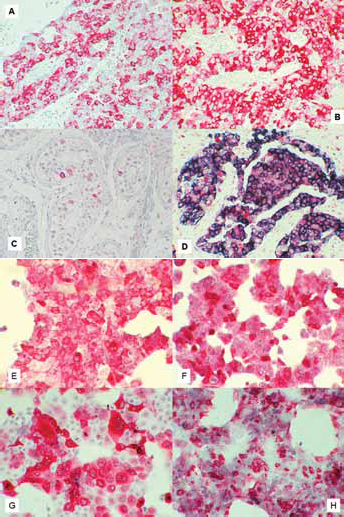
Immunohistochemical analysis of cyclin B1 and p53 expression in adult GCT and cell lines. Cyclin B1 protein expression in tumors of patient nos. 11 (A) and 8 (B). Immunostaining of cyclin B1 in testis tissue of patient no. 10, where foci of neoplastic colonization can be detected (red stain) surrounded by normal cells (C). Co-immunostaining of cyclin B1 (blue) and p53 (red) in the tumor of patient no. 2 (D). Immunoreactivity of cyclin B1 in 1411HP (E), 2102EP (F), JAR (G) and JEG-3 (H) cell lines. The embryonal carcinoma cell lines 2102EP and 1411HP differ in their sensitivity to cisplatin (sensitive and resistant, respectively). The photomicrographs in each panel were taken at ×400 magnification.
Of the 18 patients, only 1 displayed primary chemoresistance and only 3 had a partial remission (Tab. I). Thus, cyclin B1 overexpression would not seem to be associated with chemoresistance. Cyclin B1 overexpression was also independent of histology and IGCCCG stage (Tab. I). The placental choriocarcinoma (JAR, JEG-3) and testicular embryonal carcinoma (cisplatin-sensitive 2102EP and cisplatin-resistant 1411HP) cell lines all stained clearly positive for cyclin B1 (Fig. 1E-H). In particular, no difference was found between the cisplatin-sensitive and cisplatin-resistant cell lines. As reported elsewhere (9), the MCF-7 control cells showed low levels of cyclin B1, while human fibroblasts were negative (data not shown).
Unlike normal cells, where cyclin B1 is expressed at very low, almost undetectable levels in the nucleus, most of the overexpressed protein was localized in the cytoplasm of GCT cells, mirroring the typical pattern originally described in other tumor cell types (9, 15).
Cyclin B1 is overexpressed by human GCTs independently of p53 function
A direct correlation between nonfunctional p53 and cytoplasmic overexpression of cyclin B1 has been reported in human tumors (9). In our study, all 18 tumors and all 4 GCT cell lines evaluated by immunohistochemistry showed nuclear expression of p53 (mean 80%; range 50%-90%) (Tab. I), in agreement with previous reports (2, 16). There was no evidence of p53 cytoplasmic sequestration in any tumor. Both p53 and cyclin B1 expression was observed in the embryonal carcinoma (2102EP and 1411HP) and placental choriocarcinoma (JAR and JEG-3) cell lines examined by Western blot analysis (Fig. 2; data not shown). The MCF-7 cell line is known to harbor low levels of wild-type p53 and to weakly express cyclin B1 (9), and was therefore used as control.
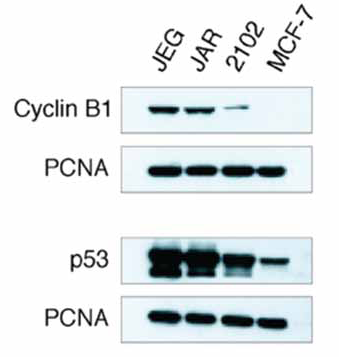
Cyclin B1 and p53 protein in cell lysates of GCT cell lines. Expression of cyclin B1 and p53 was detected by specific mAbs in 1 testicular GCT cell line (2102EP) and 2 placental choriocarcinoma cell lines (JEG-3 and JAR). The MCF-7 breast cancer cell line was selected as an example of a cell line showing low cyclin B1 expression. PCNA was used as loading control.
p53 exons 5 to 9, where more than 95% of the mutations can be found, were sequenced from 10 tumor samples for which sufficient material was available, revealing wild-type p53 in all tumors (Tab. I). In agreement with a previous analysis of a large series of unselected GCT specimens that reproducibly demonstrated a low incidence of p53 mutations (2), the vast majority of the 18 samples we analyzed harbored wild-type p53. Moreover, cyclin B1 overexpression was detected in JAR cells carrying wild-type p53 and in JEG-3 cells harboring a point mutation in 1 allele of the p53 gene (17). Finally, as wild-type p53 may be relatively nonfunctional, the cisplatin-sensitive 2102EP and cisplatin-resistant 1411HP cell lines previously reported to express functional p53 (13) were analyzed and found to overexpress cyclin B1.
Cyclin B1 siRNAs reduce the cyclin B1 protein level but only slightly inhibit JAR and JEG-3 cell proliferation
To investigate whether cyclin B1 downregulation affects tumor cell proliferation, we transfected placental choriocarcinoma JAR and JEG-3 cells with 10 nM of siRNA targeting cyclin B1. Western blot analysis was performed 48 hours after transfection. Compared to control cells or cells transfected with control siRNA against GFP, the cyclin B1 protein levels in treated cells were clearly reduced (Fig. 3A).
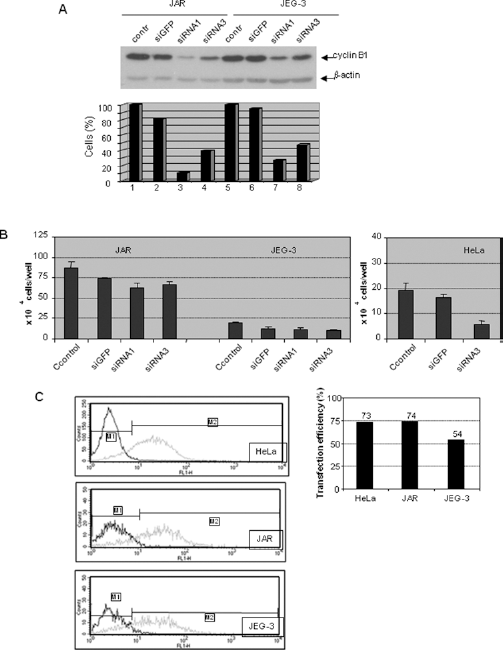
siRNAs against cyclin B1 reduced cyclin B1 protein and slightly inhibited JAR and JEG-3 cell proliferation. (A) Upper panel: Western blot analysis: the placental choriocarcinoma JAR and JEG-3 cells were treated with cyclin B1 siRNA1, siRNA3, GFP siRNA (siGFP) or transfection reagent oligofectamine alone (control) for 48 hours. Cell extracts were prepared with specific antibodies against cyclin B1 and analyzed by Western blot. β-actin served as equal loading control. Lower panel: quantification of Western blots normalized to the level of β-actin. (B) Proliferation assay: cells were treated as in (A) and were counted at 48 hours. Data are from triplicate experiments. (C) Transfection efficiency analysis. Cells were incubated with 20 nM of FITC-labeled siRNA for 6 hours, after which positive cells were determined using a Becton Dickinson FACScan flow cytometer (left panels). The percentages of positive transfected cells are shown in the right panel.
siRNA1 was more effective than siRNA3 in reducing cyclin B1 expression in JAR and JEG-3 cells. However, unlike control treatment with GFP siRNA, transfection with siRNA1 or siRNA3 reduced cell numbers by only 16% and 11% in JAR cells, and by 17% and 9% in JEG-3 cells at 48 hours, respectively (Fig. 3B, left panel). Cervical carcinoma HeLa cells, which exhibited strong proliferative inhibition after cyclin B1 downregulation in previous studies (10, 11), were used as control to explore why cyclin B1 knockdown did not efficiently inhibit the proliferation of JAR or JEG-3 cells. In contrast to JAR and JEG-3 cells, siRNA3 targeting cyclin B1 induced, as expected, strong inhibition of proliferation in HeLa cells (<10% of cyclin B1 detectable) with respect to control cells (Fig. 3B, right panel, and data not shown). Extending the incubation to 72 hours neither increased the inhibitory effect nor induced a clear G2/M arrest in JAR and JEG-3 cells (data not shown). In an effort to understand why cyclin B1 siRNA worked differently in these tumor cell lines, we labeled siRNA with a fluorescent dye (FITC) and investigated the transfection efficiency in HeLa, JAR and JEG-3 cells. As shown in Figure 3C, 54% of JEG-3 cells showed uptake of cyclin B1 siRNA compared to more than 70% of HeLa and JAR cells, indicating that transfection efficiency is not entirely responsible for the difference in HeLa, JAR and JEG-3 proliferation induced by siRNA treatment.
Discussion
Cyclins are cell cycle regulators: cyclins D1-3 and E regulate progression through G1 into S phase, while cyclins A and B affect S phase and mitosis, respectively. Whilst cyclin D2 has been consistently found to be overexpressed in GCTs (17), overexpression of cyclin E and A1 has also been reported (14, 18, 19).
There is growing interest in cyclins and CDKs as therapeutic targets in GCTs (20). Recently, a selective CDK4/6 inhibitor, PD0332991 (palbociclib), showed activity in the treatment of 1 case of relapsed nonseminomatous GCT and 3 cases of growing teratoma syndrome (21, 22).
Cyclin B1 is involved in the transition from G2 to M phase of the cell cycle (6), a checkpoint controlled by p53, which can halt the cell cycle progression at this level by lowering cyclin B1 expression (7). It has been hypothesized that cyclin B1 overexpression and the constitutive activation of cyclin B1-associated cdc2 kinase may override the p53-mediated G2/M arrest (23, 24). Cytoplasmic overexpression of cyclin B1 has been reported in several solid tumors (9, 15) and has been shown to be inversely correlated with functional p53 (9). Although p53 gene mutations are infrequent in GCT and have been correlated with chemoresistance, p53 overexpression is not uniformly found in this tumor and its mutation does not automatically correspond with resistance (2-4). In our study, cyclin B1 cytoplasmic overexpression was detected in all GCT specimens and cell lines tested, independently of chemosensitivity or p53 mutations. It has been suggested that the wild-type p53 observed in some GCTs may be relatively nonfunctional (3). However, all tumors displayed nuclear staining for p53 with no cytoplasmic sequestration, indicating the presence of functional p53. Although no definitive conclusions about p53 function in our tumor specimens can be drawn, we can confirm that the 2 embryonal carcinoma cell lines did show functional p53 expression based on response to DNA damage, as documented by other authors (13).
The incidence of GCT is increasing in Western countries (1). Although it is a curable malignancy, about 20%-25% of patients with advanced disease will relapse after first-line chemotherapy, and only 30% of these will be cured (1, 4). Thus, exploring new molecular biomarkers (including those related to p53 function) as potential targets for salvage therapy is an important challenge (25-28). Given its consistent overexpression in GCTs, cyclin B1 could represent one such therapeutic target. Deregulated cyclin B1 expression has been linked to tumorigenesis, suggesting that this molecule may be indispensable for tumor growth (29, 30), reduced protein levels, and cell proliferation (31, 10). Although the cyclin B1 protein levels in our study were clearly lower (but not suppressed) after treatment with siRNAs, the proliferation rates of JAR and JEG-3 cells were only slightly reduced. It should be noted, however, that the cyclin B1 levels were much higher in JAR and JEG-3 cells than in other tumor cells, i.e., MCF-7 (Fig. 2) and HeLa (10), where cyclin B1 protein levels were suppressed and proliferation was inhibited by the same siRNA (Fig. 3B, right panel). We compared the transfection efficiency in these cell lines (Fig. 3C), observing a lower uptake of siRNA by JEG-3 cells and a similar higher efficiency of JAR and HeLa cells with a strong inhibition of proliferation (Fig. 3B, right panel). Data from the literature indicate that up to 80% suppression of the cyclin B1 protein level does not significantly influence the growth rate of JAR or JEG-3 cells, suggesting that the amount of cyclin B1 remaining after siRNA treatment is still enough to sustain cell proliferation. The transfection of cells with plasmids encompassing short hairpin RNA targeting cyclin B1 (11) would seem to lead to continuous suppression of protein expression and may thus also impact cell proliferation. An alternative explanation is that cyclin B1 downregulation in JAR and JEG-3 cells, where p53 is active, mainly induces cell cycle arrest with little apoptosis, whereas cyclin B1 depletion in HeLa cells, where p53 is inactive due to the E6 protein encoded by human papillomavirus (32), results in cell cycle arrest accompanied by strong induction of apoptosis. Cyclin B1 knockdown may thus have to combine with other agents, which activate apoptosis pathways, to efficiently inhibit JAR and JEG-3 proliferation. It should also be borne in mind that JAR and JEG-3 cell lines derive from gestational trophoblastic disease and may therefore differ from testicular GCTs in their sensitivity to treatment with siRNA targeting cyclin B1. Interestingly, the cyclin B1 protein levels in 2102EP cells were lower than those in JAR and JEG-3 cells when assessed in parallel by Western blot (Fig. 2).
Considering the potential limitations of targeting cyclin B1 with siRNA, an alternative approach could be to inhibit CDK1, the catalytic subunit of CDK1/cyclin B1, in GCTs. CDK1 has been hypothesized to be a promising candidate for antitumor therapy by both academia and the pharmaceutical industry. Small molecule compounds inhibiting CDK1 activity are well documented, including a prominent inhibitor, flavopiridol (32). Finally, cyclin B1 fulfills several requirements of tumor-associated antigens, such as spontaneous presentation of peptides by tumor cells (9) and availability of specific T cells in the host repertoire (9, 15). However, some hurdles may hamper its recognition by CD8+ T cells in GCT, e.g., lack of expression of HLA class I molecules (33). On the other hand, anti-cyclin B1 circulating antibodies have been detected in patients with different types of cancer in which cyclin B1 is overexpressed (34, 35), but also in healthy individuals, highlighting the absence of both B-cell tolerance to this self protein and harmful effects on healthy tissues (36). Furthermore, anti-cyclin B1 vaccination was shown to protect mice from a challenge with a p53(-/-) mouse tumor cell line overexpressing cyclin B1 and to delay spontaneous cyclin B1(+) tumor growth in p53(-/-) mice (36).
In conclusion, GCTs consistently overexpress cyclin B1. Unlike in the majority of other tumors, in GCTs the overexpression is associated with the presence of functional p53.
Footnotes
Acknowledgments
The authors thank Dr. Thomas Mueller (Halle, Germany) for the generous gift of the 2102EP and 1411HP cell lines, Andrea Krämer for her excellent technical assistance, and Ursula Elbling for editing the manuscript.
Financial support: This work was partly supported by grants from Oncologia Ca’ Granda ONLUS (OCGO), Associazione Italiana Ricerca Cancro (AIRC), Milan, Italy, and Deutsche Krebshilfe #107594 (for JY and KS).
Conflict of interest: None of the authors have any financial interest related to this study.