
Abstract
Prostate cancer is the most common type of cancer in men. The antibody-mediated therapy for cancer treatment depends on the identification of selected molecular targets. The prostate-specific membrane antigen (PSMA) is a potential molecular target in prostate cancer and is abundantly expressed in this type of cancer. This study is aimed at designing and producing a recombinant PSMA epitope and a monoclonal nanobody with a high affinity toward the PSMA protein. A DNA fragment encoding the dominant epitopes of PSMA was designed, synthesized, and expressed in E. coli BL21 (DE3). A camel was immunized with the purified recombinant PSMA (rPSMA). Following mRNA isolation and cDNA synthesis, the variable fragment of heavy-chain antibodies (VHH) fragments were cloned and displayed on the surface of an M13 phage and used in sequential panning rounds. After phage ELISA and selection of colonies with the highest affinity, soluble nanobodies were produced and evaluated. Affinity of the nanobodies to rPSMA was estimated to be 3.5 × 10−7. Adherence of the purified anti-PSMA VHH was tested in cell-ELISA in the LnCaP and PC3 cell lines. VHH efficiently bound to LnCaP cells. The high specificity and affinity of this nanobody suggests its possible application as an effective tool in the diagnosis and treatment of prostate cancer.
Introduction
Prostate cancer is the most common form of cancer in industrialized countries, and represents the third leading cause of cancer-related deaths (1–3). Early diagnosis of prostate cancer can lead to successful treatment (3) by targeting specific markers in prostate cancer cells (4). The prostate-specific antigen (PSA) has been used to diagnose prostate cancer and identify tumor progression, and, in its most controversial role, to screen for prostate cancer. Recently, early diagnosis and management of prostate cancer has been revolutionized, and much has been learned about the strengths and weaknesses of this assay. Studies are still under way to definitively determine whether PSA screening makes any real difference in the detection of this disease and in subsequent patient survival.
In contrast to PSA, which is a secretory protein, the prostate-specific membrane antigen (PSMA) is a type II membrane glycoprotein with a molecular weight of about 100 kDa (2, 5, 6). PSMA is considered to be an excellent prostate tumor cell marker. PSMA expression is primarily prostate-specific, with very low levels detected in the brain, salivary glands, and small intestine. Since PSMA expression increases 10 folds in most prostate cancer cells (2, 7) it was selected as a target for an FDA-approved prostate cancer-imaging agent (111In-labeled 7E11 monoclonal antibody in Prostascint) (8, 9). In addition, there are various monoclonal antibodies that bind a specific epitope in the extracellular domain of PSMA (6, 8). Because PSMA expression is mostly limited to the prostate tissue, it has been considered to be a great target for prostate cancer therapy. Importantly, this protein is expressed at all stages of the disease, is expressed exclusively on the cells' surface, and cannot be found in blood (1, 3, 10, 11).
PSMA is known to be a glutamate carboxypeptidase II (EC 3.4.17.21), or folate hydrolase (3, 6), composed by 3 parts: a small intracellular fragment (amino acids 1-18), a signal transmembrane segment (amino acids 19-43), and an extracellular region (amino acids 44-750) (1, 10, 12). The extracellular part of PSMA folds into 3 different domains: domain I or protease domain (amino acids 57-116 and 352-590), domain II or apical domain (amino acids 117-351), and domain III or C-terminal domain (amino acids 591-750) (13).
Cancer diagnosis and treatment would greatly benefit from molecules that could bind tightly and specifically to the surface of malignant cells. Antibody therapies with such characteristics are suggested for cancer treatment with a minimum toxicity (3).
New antibodies directed towards the extracellular portion of PSMA are currently being evaluated as potentially better imaging and therapeutic agents. Althoug conventional antibodies have the ability to specifically recognize tumor cell markers, their large size and immunogenicity often limit their pharmacological value. Antibody fragments, such as monovalent antibody fragments (Fab), single-chain Fv (scFv), and nanobodies (such as the variable fragment of heavy-chain antibodies, VHH), can be produced to be used as binding agents (3, 14). These antibodies have an improved diffusion in tumor tissues, better pharmacokinetics, and a lower immunogenicity (3).
Heavy-chain antibodies naturally obtained from camels and llamas lack the CH1 domain (14–16). In order to distinguish the variable domain of these heavy-chain antibodies from the classical ones, the former is called VHH while the latter is referred as VH.
VHHs are the smallest (about 120 amino acids) antibody fragments capable of binding to antigens (15). In addition to their small size, VHH are usually more stable (especially in heating conditions) and more soluble than conventional antibodies, and are capable of recognizing haptens and cryptic epitopes that are not accessible to classical antibodies (15, 17, 18). Also, VHH can be produced in microorganisms such as E. coli, Lactobacillus, P. pastoris, A. awamori, S. cerevisiae, and tobacco plant with high yields (15). VHHs can bind to their targets with a high affinity and can be produced by simple methods with high yields (16, 19). Due to the similarity of VHHs to human VHs, their use has clinical applications with minimal immunogenic reactions (15, 17). In this study we produced a high-affinity VHH against PSMA using phage display technology. A camel was immunized with the recombinant PSMA, and a VHH-phage library was then produced. After panning and screening, VHH with the highest affinity toward PSMA were selected and expressed in their soluble form. Selected VHHs were then characterized and their affinity to PSMA was evaluated.
Materials and Methods
Expression and purification of recombinant PSMA (rPSMA)
A part of the apical domain with the most recognized antigenic epitopes was selected for rPSMA production. A 690-bp sequence of the human PSMA gene, encoding amino acids 150-380, was synthesized, subcloned into the pET28a vector (Novagen), and expressed in E. coli BL21 cells. Transformed cells were grown in LB (Lauria-Bertani) medium containing 70 μg/mL kanamycin. Expression was induced with 1 mM IPTG (Isopropyl thio-β-D-galactoside) at the optical density of 0.7 at 600 nm. The rPSMA was purified by affinity chromatography using Ni-agarose column (Qiagen). The recombinant protein was identified by SDS-PAGE and confirmed by western blotting. Concentration of the recombinant protein was determined with the Bradford method (20). The purified protein was stored at −20°C until further use.
Confirmation of rPSMA by ELISA
A total of 18 wells of a 96-well microplate were coated with 10 μg of rPSMA in 100 μL of carbonate/bicarbonate coating buffer (0.1 M Na2CO3, 0.1 M NaH-CO3, pH 9.5) and incubated for 16 hours at 4°C. The wells were washed 3 times with 200 μL of PBS-T buffer (2 mM NaH2PO4, 13 mMNa2HPO4, 150 mM NaCl; pH 7.4 containing 0.05% tween-20). The wells were then blocked with 200 μL MPBS (5% milk in PBS) and incubated for 1 hour at 37°C. After washing the wells, 100 μL of anti-PSMA mouse antibody (Pasteur Institute, Iran) was added to the wells at a dilution ranging from 1/200 to 1/12,800. Two wells without antigen and 2 wells without antibody served as negative controls. After washing, 100 μL of anti-mouse rabbit antibody conjugated with horseradish peroxidase (Qiagen) were added to each well at a 1/5,000 dilution. Binding of the anti-PSMA to rPSMA was determined by adding 100 μL of tetra-methyl benzidine (TMB) as substrate. The optical density was measured at 450 nm after stopping the reaction with 100 μL of 3N H2SO4.
Immunization of the camel
The camel was immunized with a 2-mL muscular injection of 1 mg of rPSMA with an equal volume of Freund's complete adjuvant (Sigma-Aldrich). Booster injections containing 2, 3, 4, and 5 mg of rPSMA with an equal volume of Freund's incomplete adjuvant were administered at intervals of 10 days. Blood samples were collected before each injection, and sera were used to evaluate the immune response by ELISA. The ELISA was carried out as described above. Camel serum at a volume of 100 μL was added to the wells at 1/500, 1/1,000, 1/3,000, and 1/5,000 dilutions. After 1 hour of incubation and washing of the wells, 100 μL from a 1/6,000 dilution of anti-camel rabbit antibody conjugated with horseradish peroxidase (HRP) (Qiagen) were added to each well. All immunization steps and animal handling were performed according to the University's guidelines and under the supervision of a veterinarian. All procedures and ethical issues complied with relevant laws and institutional guidelines of the Shahed University.
Isolation of camel lymphocytes
A total of 200 mL of blood were taken from the cervical artery of the immunized camel 10 days after the fifth injection. White blood cells were separated by Ficoll (Sigma) discontinuous gradient: 6 mL of blood were slowly added into a tube containing 3 mL of Ficoll, and centrifuged at 2000 g for 15 minutes. Lymphocytes were washed with sterile PBS, counted, and stored at −70°C.
Isolation of RNA and synthesis of cDNA
RNA purification was performed with the high pure RNA isolation kit (Roche), according to the manufacturer's specifications, and verified by electrophoresis. RNA was then stored at −80°C. The RNA yield was measured with a Pichodrop™ (Germany) at 260 nm and 280 nm. Total RNA was used for synthesis of the first-strand cDNA with the Prime Script 1st Strand cDNA Synthesis Kit (Fermentas). An oligo dT was used as a primer and cDNA synthesis was carried out according to the manufacturer's protocol. The resulting cDNA was stored at −20°C.
Amplification of the VHH gene by nested PCR
Isolation of the VHH from the variable domain of classical antibodies (VH) was carried out by nested PCR on cDNA using the following primers:
A4 (Forward primer): 5′-GTCCTGGCTGCTCTTCTACAAGG-3′
A6 (Reverse primer): 5′-GGTACGTGCTGTTGAACTGTTCC-3′
First, the PCR reaction was carried out with an initial denaturation step at 94°C for 10 minutes followed by 30 cycles (at 94°C for 30 seconds, at 66°C for 35 seconds, and at 72°C for 35 seconds), and a final extension at 72°C for 10 minutes. Then, 600-bp and 700-bp bands (VHH-hinge-CH2) were purified with the Gel Extraction kit (Bioneer) and used as templates for the second PCR. Appropriate restriction sites (sfiI) were introduced into the following primers for cloning into the pComb3x phage display vector (Creative Biogene, USA).
Fr4-SfiI (Forward primer):
5′-ACTGGCCCAGGCGGCCGAGGTGCAGCTGSWGSAKTCKG-3′
Fr1-SfiI (Reverse primer):
5′-ACTGGCCGGCCTGGCCTGAGGAGACGGTGACCWGGGTC-3′
A PCR reaction was carried out with an initial denaturation step at 94°C for 8 minutes, followed by 30 cycles (at 94°C for 30 seconds, at 64°C for 35 seconds, and at 72°C for 35 seconds) and a final extension at 72°C for 10 minutes. The PCR product was analyzed by electrophoresis and purified with the gel extraction kit. For all PCR reactions (Biogen, Turkey) we used the Taq DNA polymerase and dNTPs.
Construction of the VHH gene library
The VHH gene and pComb3x vector were digested with the sfiI restriction enzyme (Fermentas) and purified by using the gel extraction kit. The digested VHH genes were ligated into the phagemid pComb3x by T4 DNA ligase (Fermentas). Ligation products were transferred into competent E. coli XL1 blue by electroporation. The library size was determined by plating 10 μL of the transformant on 100 μg/mL ampicillin LB agar plates. The colonies were counted to calculate the size of the library. VHH cloning was confirmed by PCR on 50 randomly selected clones as described before under “Amplification of the VHH gene by nested”.
Production of nanobody phage library
Recombinant phage particles expressing nanobodies were produced by superinfection of transformed bacteria with the M13K07 helper phage (Amersham, USA). Transgenic bacteria were grown in 30 mL of 100 μg/mL ampicillin LB broth at 37°C and 260 rpm. A total of 1012 M13K07 were inoculated at log-phase OD600 of 0.5 followed by incubation at 37°C for 1 hour (30 minutes without shaking and 30 minutes shaking at 260 rpm). A total of 200 mL of LB containing 70 μg/mL kanamycin and 100 μg/mL ampicillin were added and the culture was incubated overnight at 37°C and 260 rpm. Recombinant phages were isolated by centrifugation at 5,000g at 4°C for 10 minutes. The supernatant was collected and the phages were precipitated by addition of 20% (V/V) PEG solution (20% polyethylene glycol 6000 in 2.5 M NaCl in distilled deionized water). After incubation for 1 hour on ice, the mixture was centrifuged for 25 minutes at 15,000g at 4°C. The supernatant was discarded and the phage pellet was dissolved in 1 mL of PBS. Centrifugation was performed for 5 minutes at 12,000g at 4°C. The supernatant containing the nanobody phage library was stored at 4°C. Phage titration on phage-VHH library was carried out by infecting XL1-Blue cells and plating them on 100 μg/mL ampicillin LB agar.
Biopanning of phage-VHH against rPSMA
Two wells of 96-well plates were coated with 100 μL of 10 μg/mL rPSMA in PBS and were incubated overnight at 4°C. Wells were washed 3 times with 200 μL PBST (PBS with 0.05% tween-20) and were then blocked with 200 μL of 5% BSA (w/v) in PBS and incubated at 37°C for 2 hours. After washing the wells with PBST, 100 μL of phage-VHH library was added to each well and the plate was incubated at 37°C for 2 hours. Wells were washed 10 times with PBS containing 0.1% Tween-20. Bound phage-VHHs were eluted with 100 μL of 0.2 M Glycin-HCl, pH 2.2, at room temperature. The eluted phages were transferred to a new tube and immediately neutralized with 14 μL of 1 M Tris-HCl (pH 9.0). The eluted phages were titrated and inoculated with log-phase XL1blue bacteria in SB medium (Super Broth) and incubated at 37°C for 1 hour (30 minutes without shaking and 30 minutes shaking at 260 rpm). Ampicillin at a 80 μg/mL concentration was added to the medium and incubated at 37°C for 30 minutes. One milliliter of helper phage (1012 phage particles) was added to the medium and incubated at 37°C for 1 hour (30 minutes without shaking and 30 minutes shaking at 260 rpm). Then, 200 mL of SB containing 70 μg/mL kanamycin and 80 μg/mL ampicillin were added and incubated overnight at 37°C and 260 rpm. Phages were collected as described before. Phages were then titrated and used in sequential rounds of panning. From the second to the fifth step, biopanning was performed by increasing the tween-20 concentrations from 0.1% to 0.6%.
Analysis of biopanning by polyclonal phage ELISA
An ELISA reaction was performed to check the biopanning process. For polyclonal phage ELISA, 16 wells of 96-well plates were coated with 100 μL of 10 μg/mL rPSMA in bicarbonate coating buffer and were incubated overnight at 4°C. After washing with PBST the wells were blocked with 150 μL of MPBS and incubated at 37°C for 1 hour. Wells were washed 3 times by PBST and 100 μL of phage solution from each biopanning processes were added into 2 wells. The plate was incubated at 37°C for 1 hour. After washing the wells with PBST, 100 μL of anti-M13 antibody conjugated to HRP (Amersham) at a 1:8,000 dilution was added to each well and was incubated at 37°C for 30 minutes. The wells were washed 3 times with PBST and 100 μL of TMB were added to each well. After incubation for 15 minutes at room temperature the reaction was stopped by H2SO4 and the OD450 was measured.
Selection of phage antibodies
After 7 rounds of panning, 18 colonies of the fourth round were isolated and inoculated into 30 mL of 100 μg/mL ampicillin LB broth. Phage particles were released with helper phage as described under “Production of nanobody phage library”. Polyclonal phage ELISA against rPSMA was carried out as described under “Analysis of biopanning by polyclonal phage ELISA” and 2 of the best binding VHH-phage clones were selected for production of VHH.
Sequencing of VHH
The gene-expressing C3 nanobody in pET28a was sequenced using Fr4-SfiI and Fr1-SfiI specific primers (Bioneer Company). The result was aligned with NCBI BLAST tools.
Expression and purification of soluble VHH
E. coli Top10F' cells grown in LB medium were infected with the selected VHH-phage solution at an OD600 of 0.5. The culture was incubated at 37°C for 1 hour (30 minutes without shaking and 30 minutes shaking at 260 rpm). Cells were collected by centrifugation at 4,000 g for 15 minutes at 4°C and cultured on LB agar plate containing 100 μg/mL ampicillin and incubated at 37°C for 16 hours. A single colony was inoculated in 100 mL of SB medium containing 100 μg/mL ampicillin. IPTG at a final concentration of 1 mM was added to the culture at OD600 and the culture was incubated overnight at 28°C and 280 rpm.
For VHH purification, the bacteria were collected at 4,000g for 15 minutes and were resuspended in 3 mL of PBS containing 1 mM phenylmethylsulfonyl fluoride (PMSF) and then sonicated. The mixture was centrifuged at 15,000 g for 25 minutes at 4°C. The supernatant was loaded on Ni-agarose column (Qiagen). Serial washing was carried out with buffers containing respectively 20, 30, and 40 mM immidazole. Finally, the nanobody was eluted with the same buffer containing 250 mM imidazole. Soluble VHH was dialyzed against 10 mM phosphate buffer at pH 7.2 for 24 hours. The expression was confirmed by SDS-PAGE and Western blotting. VHH concentration was measured by spectrophotometry at 280 nm.
In order to increase the expression level, the VHH gene was subcloned into the pET28a vector with EcoRI and HindIII restriction sites. The cloning was confirmed by PCR and plasmid digestion. VHH expression and purification was carried out as described under “Expression and purification of recombinant PSMA (rPSMA)”.
Determination of the binding affinity
A clone named C3, with the highest score in monoclonal phage-ELISA was chosen for further studies. Microtiter plates were coated overnight with 100 μL of rPSMA in bicarbonate with concentrations of 0, 2.5, 5, 10, and 20 μg/mL at 4°C. The same concentration of BSA was coated as non-specific antigen (NSB). The wells were blocked with MPBS and incubated for 1 hour at 37°C. Then, wells were washed with PBST, and 100 μL of soluble VHH antibodies were added at the concentrations of 1.25, 2.5, 5, 10, and 20 μg/mL, and incubated at 37°C for 2 hours. After washing with PBST, an anti-Hemagglutinin antibody conjugated to HRP (HA-HRP-antibody), was added at a 1/10,000 dilution in PBST to each well, and incubated at 37°C for 1 hour. The wells were washed and 100 μL of TMB were added to each well and incubated for 15 minutes at 37°C. The optical density was measured at 450 nm after the reaction was stopped. The association constant (Ka) of nanobody against rPSMA was calculated using the formula described by Beatty et al in 1987 (21) to determine the VHH affinity.
Determination of binding specificity
The cross-reaction was tested with cancerous antigens, such as VEGF and CA9, and non-related proteins available in our laboratory, such as BSA, UreC (Helicobacter pylori), BoNTA (Clostridium botulinum), Bap (Acinetobacter baumannii), and LPS (Salmonella typhimurium and Vibrio cholerae). All the antigens were coated in microtiter plates at the concentration of 10 μg/mL. After washing the plates with PBST, 100 μL of a 20 μg/mL solution of the purified recombinant nanobody were added to each well and the assay was performed as described in previous sections.
Anti-PSMA nanobody efficiency in cell-ELISA
The purified anti-PSMA VHH was tested by cell-ELISA in the LnCaP and PC3 cell lines. Each cell line was cultured at 5×104 cells per well in a 96-well ELISA plate and incubated in a CO2 incubator at 37°C for 24 hours. After removal of the culture medium, the cells were fixed for 15 minutes at −20°C with acetone/methanol solution. The wells were blocked with 200 μL of 3% skimmed milk in PBS for 45 minutes at 37°C. After washing the wells with PBST, a total of 40, 20, 10, 5, and 2.5 μg of anti-PSMA VHH were added to each well and the plate was incubated at 37°C for 2 hours. The wells were then washed several times with PBST, and 100 μL of HRP-conjugated anti-His tag antibody (Qiagen) were added to the wells at a 1:40,000 dilution. After 1-hour incubation at 37o C, the wells were washed with PBST several times and 100 μL of TMB substrate were added to the wells. The reaction was stopped after 15 minutes with 3N H2SO4 and absorbance was read at 450 nm.
The same procedure was repeated with cells in suspension. Each cell line was used at 1×105 cells per reaction. After each step, cells were collected by centrifugation at 2,000g for 8 minutes at 4°C. For washing, cells were resuspended in 500 μL of PBST. All antibodies were used in a 200-μL volume, and at the same concentrations described above. In control reactions, PBS was used instead of anti-PSMA VHH. For quantification of the immunoreaction, cells were sedimented by centrifugation and the supernatant was transferred to the wells of the ELISA microplate.
Results
Expression and purification of rPSMA
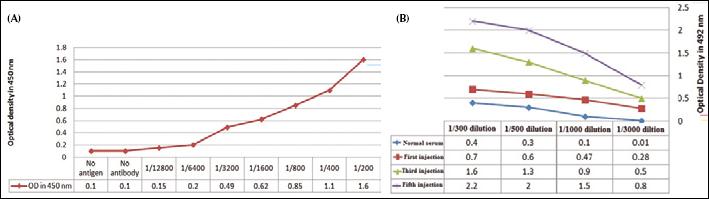
The presence of the rPSMA protein was confirmed by SDS-PAGE and Western blot analysis (Fig. 1A). The recombinant protein was purified by immobilized affinity chromatography (Ni-agarose) and analyzed by SDS-PAGE (Figs. 1B and 1C). The presence of rPSMA was also confirmed by ELISA using the hybridoma anti-PSMA antibody (Fig. 2A).
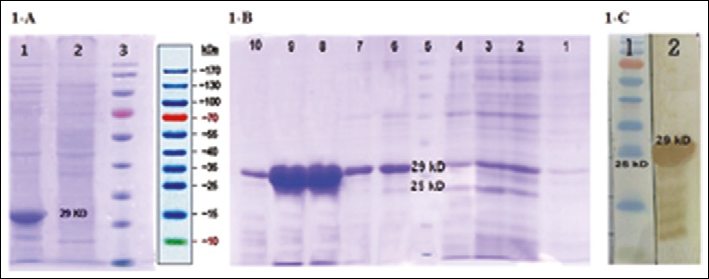
Expression and purification of rPSMA on a 12.5% acrylamide SDS-PAGE.
Immunization of the camel
The serum from the immunized camel was analyzed for antibody titer after each immunization step, and the results showed a maximum titer level after the fifth injection (Fig. 2B).
Reproduction of the VHH gene by nested PCR
RNA was isolated from the lymphocytes obtained after the fifth injection. cDNA was generated using oligo dT primers. Three bands were observed in the first PCR: the 900-bp band corresponded to the conventional antibody domain (VH+CH1+hing+CH2), and the 2 additional bands (of 620 and 690 bp) represented the non-conventional antibody domain (VHH+hing+CH2). For the amplification of the VHH genes, smaller fragments were gel-purified and used as templates in the second PCR.
Production of the nanobody phage library
The VHH fragments cloned in the pComb3x phagemid vector showed a library size of 3×106. PCR analysis revealed the presence of VHH genes in 95% of the clones.
Polyclonal phage ELISA
The phages obtained from each round of panning were used in an ELISA reaction for the detection of rPSMA (Fig. 3A). The optical absorption at 450 nm reached 1.4 in the fourth round of biopanning.
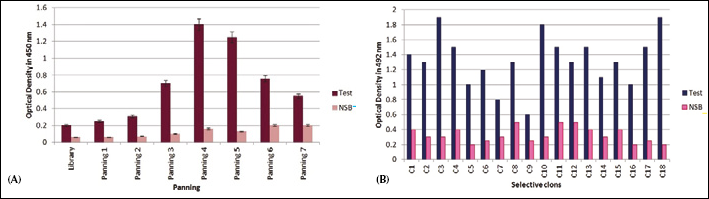
Selection of phage antibodies
For the isolation of high-affinity nanobodies against rPSMA, 18 colonies were selected from the fourth round of panning. The colonies number 3, 10, and 18 showed the highest affinity towards rPSMA (Fig. 3B). These colonies were confirmed by colony PCR and restriction analysis.
Production and purification of soluble VHH
E. coli Top 10 F' bacteria were infected with phages obtained from the clones number 3 and 18. Transformation was confirmed with colony PCR and phagemid digestion. The result of the nanobody expression is shown in Fig. 4A. On SDS PAGE the molecular weight of the nanobodies was 17 kDa. VHH purification was performed using Ni-agarose chromatography followed by analysis on SDS-PAGE and Western blotting (Figs. 4B and 4C).
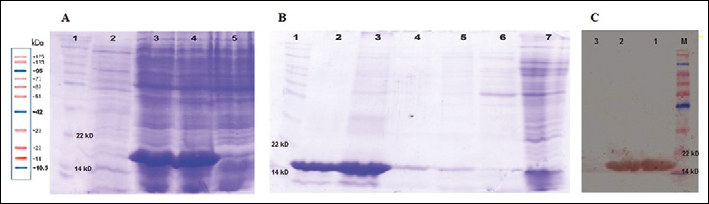
Binding affinities and specificities
The affinity of C3 calculated with the method proposed by Beatty et al (21) was 3.5×10−7 M. Among several proteins or antigens used in ELISA, rPSMA scored the highest optical density in the cross-reaction assay (Fig. 5).
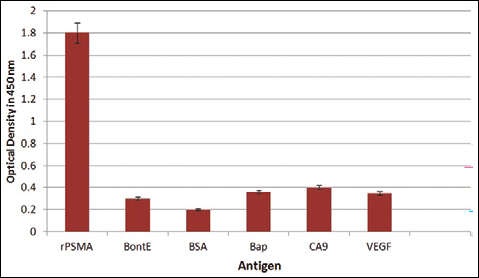
Anti-PSMAVHH specificity was tested by ELISA and compared with other cancerous antigens, such as VEGF and CA9, and non-related proteins, such as BSA, UreC (Helicobacter pylori), BoNTA (Clostridium botulinum), Bap (Acinetobacterbaumannii), and LPS (Salmonella typhimurium and Vibrio cholerae).
Anti-PSMA nanobody efficiency in cell-ELISA
Anti-PSMA VHH could efficiently attach to the expressed PSMA protein on the surface of LnCaP cells, whereas no immunoreaction was observed when applying anti-PSMA VHH on PC3 cells (Fig. 6). In the suspension form, the anti-PSMA VHH showed a 150% higher immunoreaction than that in PC3 cells.
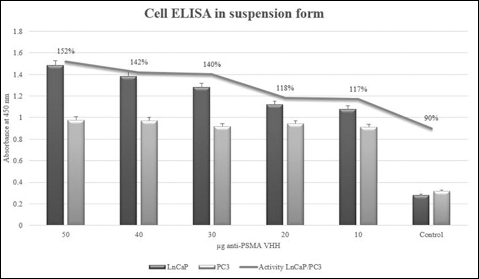
Anti-PSMAVHH binding activity in the LNcaP and PC3 cell lines. Binding of the anti-PSAMVHH was studied in 1×105 cells of each LNcaP and PC3 cell line in suspension. An amount of 50 μg of anti-PSMAVHH showed 150% of more activity in LnCaP cells than in PC3.
Sequencing of VHH
The VHH gene was for the 90% similar to that of Camelus dromdarius IgVH.
Discussion
PSMA expression is 10-fold increased in prostate cancer (2, 22). A significant correlation between PSMA expression and aggressiveness of the disease highlights the importance of PSMA in prostate cancer (12). Since in specific conditions, such as antibody binding, PSMA is endocytosed, this antigen is considered as an excellent target for immunotherapy. This characteristic can be used for cancer detection and tissue-specific delivery of various therapies (9, 23). Because the PSMA expression level is higher in prostate cancer than in normal tissues or benign prostatic hyperplasia (BPH), it has a distinctive advantage over other prostate cancer markers (7, 24).
The PSMA extracellular region consists of a stretch of amino acids from 44 to 750. A portion of the epithelial surface of PSMA was selected bioinformatically and several antigenic domains were identified within this region. Also, the region underwent bioinformatics analysis for predicting B-cell epitopes. Finally, paratope domains of known PSMA antibodies, such as J591, J415, J533, and E99 (25), were mapped on the extracellular sequence. In order to maintain a natural folding of the PSMA protein, the region containing most of the dominant epitopes was selected instead of constructing a chimerical protein from these epitopes. The selected region consisted of amino acids from 150 to 380. The lack of disulphide bonds and proper size of this recombinant protein facilitated its expression in prokaryotic systems such as E. coli. Since PEQ226.5 (an anti-PSMA antibody) is able to detect the antigen (12), Lana et al (12) suggested that that PSMA epitopes recognized by the PEQ226.5 antibody were located between amino acids 134 and 437. This selected segment includes a part of the domain II of PSMA (amino acids 117 to 351). Mindy et al (6) established that the epitopes recognized by the J591 anti-PSMA monoclonal antibody are located between amino acids 153 and 374. This area is also localized in domain II (6).
Antibody treatment is a suitable approach for cancer therapy (3). Considering the better pharmacokinetic properties of smaller ligands as well as their decreased non-specific uptake and immunogenicity, for our purpose we decided to use VHH, which is smaller than the conventional antibodies and has better tissue penetration. Nanobodies can easily be produced in prokaryotes such as E. coli (18, 26, 27). The main characteristics distinguishing VHH from other antibody fragments are its simple cloning procedure to obtain a single gene, and thus its easy selection from libraries (19, 28). The single chain of VHHs makes these antibodies suitable for their use in display systems such as phage display, a powerful tool for antibody detection and isolation. Approximately 30% of human antibodies used for treatment have been produced by phage display (26, 27). The human anti-mouse antibody (HAMA) response is one of the main problems associated with antibodies produced by the hybridoma technique.
In addition, there is no control over the antibody affinity. In the phage display technique, the process can be optimized based on the selection of high affinity antibodies (28, 29). To avoid the isolation of VH genes, we used nested PCR. This approach can lead to mutagenesis and molecular diversity, which not only leads to library diversity but also prevents the contamination of the library with VH genes (15). A comprehensive study was carried out on all nanobody genes registered at the NCBI databases. As previously reported (18, 30–32) the second primers were designed in degenerated forms to allow the maximum isolation of VHH genes.
In order to ensure an efficient recognition of the nanobody and its binding to the PSMA protein naturally expressed on the cell surface, the nanobody was tested in a cell-ELISA with the LNcap and PC3 cell lines. Except for the ability to express the PSMA protein, PC3 cells are morphologically identical to LnCaP cells. Therefore, any deference in ELISA immunoreactivity would be due to the binding of anti-PSMA VHH to the PSMA expressed on the surface of LnCaP cells. This difference was observed in a cell-based ELISA performed with LnCaP and PC3 cell lines using the obtained VHH as a primary antibody. The higher immunoreactivity of LNcap cells compared to PC3 cells clearly confirms the efficiency of VHH to bind to the natural PSMA protein and recognize PSMA-expressing cells. This assay seems thus to be important for prostate cancer diagnosis and therapy. The absence of considerable immunoreaction in PC3-coated wells and empty, blocked wells indicates the lack of VHH affinity to normal cells. The ability to distinguish normal cells from cancerous cells is vital in cancer diagnosis and treatment. Nevertheless further tests are needed to establish the clinical applicability of the anti-PSMA VHH. The results are promising for VHH nanobodies in terms of high tissue penetration, low immunogenicity, and specificity. The isolated anti-PSMA VHH showed a high affinity and a successful binding to PSMA-expressing LnCaP cells, and no binding to PC3 cells. Despite the good results in terms of high specificity and affinity suggests the possible application of this nanobody for the diagnosis and treatment of prostate cancer, further confirmations are needed. Specifically, studies dealing with antibody maturation and labeling evaluating the possible cross-reactions and efficiency of in vivo tumor recognition in animal models are required for its therapeutic application.
Footnotes
Acknowledgment
The authors would like to thank the Biotechnology Development Council of I. R. Iran and Shahed University, Tehran, Iran, for supporting this study