
Abstract
The detection of somatic mutations in a tumor represents a valuable tool for tumor characterization and provides the clinicians with information for setting up the most appropriate therapy. KRAS mutations in codons 12 and 13 are important biomarkers routinely analyzed in the clinic for the management of anti-EGFR treatment in colorectal carcinoma (CRC).
Here we report a sensitive and inexpensive assay for KRAS mutations based on a PNA-mediated PCR clamping. The assay displays very high sensitivity (0.7%) and specificity (96.7%) when compared to traditional sequencing (SS) and pyrosequencing (PS), two of the most commonly and routinely used methods employed today by diagnostic laboratories. Furthermore, the PNA assay requires only basic and low-cost laboratory equipment, in contrast with all the most recent PCR-based technologies, which are highly sensitive but also much more expensive.
Finally, despite the PNA assay does not allow for the definition of specific mutations, it is the cheapest and easiest screening method to firstly stratify wild-type and mutated patients, information that is strictly necessary to clinicians for the management of CRC and anti-EGFR treatment.
Introduction
The KRAS proto-oncogene is a crucial downstream molecule in the EGFR pathway, controlling pivotal intracellular signals including cell growth, differentiation, and apoptosis (1).
Mutations of KRAS, leading to uncontrolled activation of RAS effectors, occur prevalently within codons 12 and 13 (up to 98%), and are often detected in different cancer types including lung, pancreatic, and colorectal cancer (CRC) (2, 3). In clinical practice an early and accurate detection of KRAS mutations is an essential prerequisite for choosing the most appropriate therapy and the use of anti-EGFR monoclonal antibodies (cetuximab and panitumumab) (4–6). Sensitivity and specificity of mutations’ detection assays might be influenced by poor DNA quality and scarce number of tumor cells (7). Although the Sanger sequencing (SS) is widely accepted to date as the gold standard method for mutations’ detection, it is time-consuming, cost-ineffective, and reaches a sensitivity of 15%-20% (8), This latter point represents a great disadvantage due to the high heterogeneity of tumor tissues. Despite pyrosequencing (PS) seems to be a promising alternative with an improved detection limit (5% sensitivity), a relatively fast run time, and the ability to easily quantify the amounts of a mutant allele (9, 10), its availability still remains prerogative of only few centers, due to its high cost. Moreover, 5%-10% of samples fall into a “grey area” in which genotyping is not able to strongly support clinicians in their therapeutic decisions.
The availability of a simple, more sensitive, and quick assay allowing a rapid and unambiguous detection for KRAS mutations is clearly needed for the clinical management of CRC. In this contest, we developed an easy and user-friendly assay for the detection of KRAS mutations based on a primer exclusion PNA-directed PCR-clamping (PNA assay). Here we discuss benefits and costs, comparing it with the 2 main currently available tests (SS and PS) used in routine diagnostic laboratories on paraffin tissues collected by pathologists at diagnosis.
Materials, Methods, and Results
We here describe a new strategy for KRAS mutations’ analysis in CRC, based on a primer exclusion PNA-directed PCR-clamping (PNA-assay). Peptide nucleic acids (PNAs) are synthetic molecules with peptidic-replacing phosphodiesteric-bonds, acting as potent DNA mimic in terms of specific hybridization (11). As a consequence, the PNA/DNA heteroduplex is thermally more stable than the DNA/DNA homoduplex and a single base mismatch strongly destabilizes the PNA/DNA heteroduplex (12). The PNA-assay consists of a modified single-step PCR that makes use of a PNA probe spanning codons 12 and 13 (exon 2) of the KRAS gene. The PNA clamp molecule and KRAS forward primer were designed complementary to the wild type (wt) KRAS sequence, thus leading to a direct competition towards the complementary template genomic DNA. As a consequence, in wt samples, the PNA/template hybridization is thermodynamically favored and DNA amplification thereby suppressed. By contrast, in case of any type of KRAS mutations in codons 12 and 13, a single base-pair mismatch will strongly impair the PNA/template stability, favoring PCR amplification (Fig. 1A). Optimal experimental conditions were established for formalin-fixed paraffin-embedded specimens (containing more than 75% of tumor cells in each sample) from CRC patients.
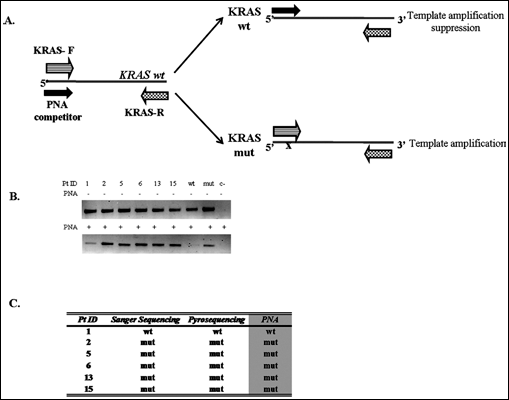
PNA-mediated clamping PCR. (A) Schematic representation of the PNA-mediated clamping PCR for the detection of KRAS mutations at codons 12 and 13 with relative positions of PCR primers (forward, KRAS-Fwd and reverse, KRAS-Rev) and PNA competitor. KRAS wt, KRAS sequence wild-type; KRAS mut, KRAS sequence mutated at codons 12/13. (B) Representative results from 6 patients analyzed for KRAS mutation by PNA assay (lower panel); positive (mut), negative (wt), and no template (c-) controls are included. For patients’ ID refer to Supplementary Material Table I. For each sample, a PCR reaction without PNA molecule (upper panel) is used as internal control of amplification. (C) Summary of mutational analysis results by comparing the 3 methods; Pt ID: patients identification number.
A total of 10 ng of genomic DNA were enough to assure fragment amplification; the single-step PCR was modified by adding an additional step at 70°C to guarantee specific PNA annealing. Discrimination between the wt and mutated alleles was performed by simply running the PCR products on a 2% agarose gel and checking for the presence of an amplification band. In order to avoid false negative results due to poor DNA samples’ quality, the results were always compared with those obtained from PCR reactions performed under standard conditions without PNA clamp molecule. Representative results from 6 patients are summarized in Figures 1B and 1C.
We urged to assess the assay sensitivity by testing serial dilutions with plasmids carrying either the wt or mutated KRAS cDNA sequences. Mutant DNA, obtained by cloning PCR amplicons of an heterozygous patient carrying the mutation GGT>GAT at codon 12, was diluted with its wt counterpart, at different percentages ranging from 100% to 0.7%. Remarkably, the analysis by PNA assay showed that mutations were reproducibly detectable in 3 independent experiments, reaching a sensitivity of mutated allele as high as 1.4% (mutated versus wt) (p<0.005) and indeed a 0.7% dilution (p=0.03) (Supplementary Methods and Supplementary Fig. 1).
Since the assay herein reported may represent a valid revolutionary tool in routinely CRC management, we analyzed by PNA assay 68 specimens collected at diagnosis and previously genotyped by both SS and PS (Supplementary Tab. I). The results showed that the PNA assay displays a sharply improved sensitivity when compared to both SS (as already reported by Kwon et al. [13]) and PS, allowing the detection of the mutated allele with a sensitivity approximately 7 times higher (0.7% for PNA assay vs 5% for PS). Genotyping showed a concordance of 60.29% among the 3 methods, the major discrepancy being the mutations identified by PNA assay that were previously missed either by SS or PS (Figs. 2A and 2B). In fact, among the 24 tumors with wt KRAS assessed by SS, PS identified 7 further mutations. In addition to those 7 mutations, the PNA assay correctly identified other 17 samples. Remarkably, the PNA assay succeeded even where PS failed. Moreover, the analysis by PNA assay of 4 samples fallen into the grey area by PS, in fact, identified the presence of KRAS mutations. Moreover, the PNA assay failed to detect KRAS mutations only in 1 case out of 30 (which resulted mutated by PS), thus displaying a specificity as high as 96.7% (calculated as the number of mutated samples identified by both PNA and PS/number of mutated samples identified by PS, excluding samples falling into the grey area by PS).
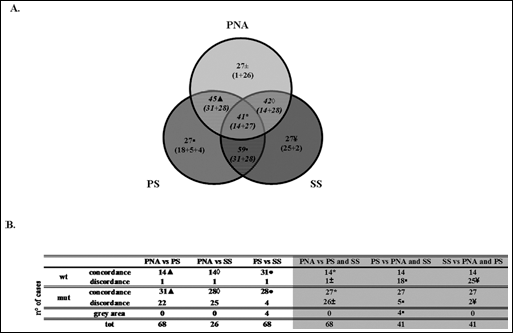
Comparison of KRAS mutational results among PNA-mediated clamping PCR, pyrosequencing, and Sanger sequencing. (A) Comparison of KRAS mutational results among the 3 methods (PNA: PNA assay; PS: Pyrosequencing; SS: Sanger Sequencing). Striped areas represent concordance, while clear areas represent discordance among methods. Symbols (▲, ⋄, ⊠, *, ±, ⊠, ¥) and numbers refer to the summarizing Table presented in Figure (B).
Discussion
In this study we describe the development of a very sensitive and specific strategy for the detection of KRAS mutations based on a PNA-mediated PCR clamping. The main objective of this work was to compare, in terms of sensibility and specificity, the newly proposed method with SS and PS, two of the most commonly and daily-used techniques applied in clinical laboratories and pathology departments. When choosing a clinical routine method, the factors to be considered, besides sensitivity and specificity, include also turnaround time and costs (9). The method here proposed shows several advantages, such as fast run time (2.5–3 hours), low costs (less than 2 Euros/sample versus 10 Euros/sample for PS), and suitability on paraffin-embedded tissues (the biological material commonly used for KRAS mutations’ determination), thus representing a very widely applicable method. Moreover, the PNA assay demonstrates a higher sensitivity (0.7%) compared to SS (20%) and PS (5%), and a specificity of 96.7%.
The most recent technologies used for the detection of KRAS mutations include next generation sequencing (NGS), Taqman PCR, CAST PCR, HRM PCR, Taqmelt PCR, as well as peculiar methods such as the BEAming assay, Intplex, and droplet dPCR (for a revision see Supplementary Tab. II). All of these methods show a high sensitivity but are very expensive, require highly qualified laboratory technicians and, right now, are prerogative of research laboratories; on the contrary, the PNA assay only requires a regular thermal cycler and an electrophoresis apparatus, all equipment widely available both in clinical laboratories and in pathology departments.
Although the PNA assay does not allow defining specific mutations, we consider this method as a reproducible, firm, and adequate strategy to discriminate, as first screening, wt from mutated patients. This will allow: (i) the clinicians to decide for anti-EGFR therapies in short time; (ii) the laboratories to save time and money (important especially if we consider the increasing number of cases analyzed every day and the concomitant heavy reduction of both human and economical resources in our public health system); (iii) the patients to benefit of a targeted therapy, thus avoiding toxicities and gaining in quality of life.
Footnotes
Acknowledgments
We thank Dr. Francesca Cavicchioli for helping in the cloning experiments.