
Abstract
Breast cancer is a highly heterogeneous disease. Tamoxifen is a selective estrogen receptor (ER) modulator and is mainly indicated for the treatment of breast cancer in postmenopausal women and postsurgery neoadjuvant therapy in ER-positive breast cancers. Interestingly, 5-10% of the ER-negative breast cancers have also shown sensitivity to tamoxifen treatment. The involvement of molecular markers and/or signaling pathways independent of ER signaling has been implicated in tamoxifen sensitivity in the ER-negative subgroup. Studies reveal that variation in the expression of estrogen-related receptor alpha, ER subtype beta, tumor microenvironment, and epigenetics affects tamoxifen sensitivity. This review discusses the background of the research on the action of tamoxifen that may inspire future studies to explore effective therapeutic strategies for the treatment of ER-negative and triple-negative breast cancers, the latter being an aggressive disease with worse clinical outcome.
Keywords
Introduction
Breast cancer is a highly heterogeneous disease based on its pathological features and can be both slow growing with good prognosis and highly invasive with poor prognostic and predictive features. 1 Dysregulation of estrogen receptor (ER) expression is observed in 60-70% of breast cancer patients.2,3 Estrogen is directly involved in cell growth via regulation of the expression of ER target genes in different tissues, including the breast. Tamoxifen, a nonsteroidal triphenylethylene derivative, is known to compete with estrogen to inhibit ER activity associated with tumor cell growth. It is a selective estrogen receptor modulator (SERM) with antiestrogenic activity in breast tissues and agonistic activity in the bone, whereas it has mixed actions in the uterus. 4 Tamoxifen was approved for the treatment of postmenopausal women suffering from advanced breast cancer by the U.S. Food and Drug Administration in 1977 and later for postsurgery adjuvant treatment to eradicate micrometastasis from primary breast cancer. Therefore, tamoxifen gradually became a mainstay hormone therapy in preventing the relapse of ER-positive cancers.4,5
The antiestrogenic activity of tamoxifen mediated by ER is well established and is the main reason for tamoxifen treatment in ER-positive breast cancers. ER-negative tumors are characterized by the lack of ER expression or expression of very small levels of ER, depending on the measurement of ER status by different laboratories.6–8 In the past, many clinical studies involving tamoxifen treatment in women with ER-negative breast cancers had not produced significant benefits in terms of mortality reduction or reduction of recurrence of breast cancer in these patients.9,10 However, new interest in investigating the mechanism of action of tamoxifen was prompted by the observations of development of resistance to tamoxifen in many subgroups of ER-positive breast cancer patients, depending on the expression of various molecular markers, presence of estrogen receptor subtype beta (ERβ) in ERα- and/or progesterone receptor (PR)-negative breast cancers, improvement of prognostic outcomes in ER-negative patients with ERβ expression upon tamoxifen treatment, and discovery of several ER-independent mechanisms in ER-negative breast cancer cells treated with tamoxifen.11–18 Overall, the response to tamoxifen in very small cohorts of ER-negative patients led to further research into the action of tamoxifen on ER-negative breast cancer cells to explore the molecular mechanisms underlying this phenomenon.
In this review article, we explore the ER-subtype-dependent effects of tamoxifen treatment, the role of ER-negative subtype status, and variations in tumor microenvironment and deregulated epigenetic mechanisms that may contribute to the modulation of tamoxifen sensitivity in ER-negative breast cancers. These mechanisms of tamoxifen action are depicted in Figure 1.
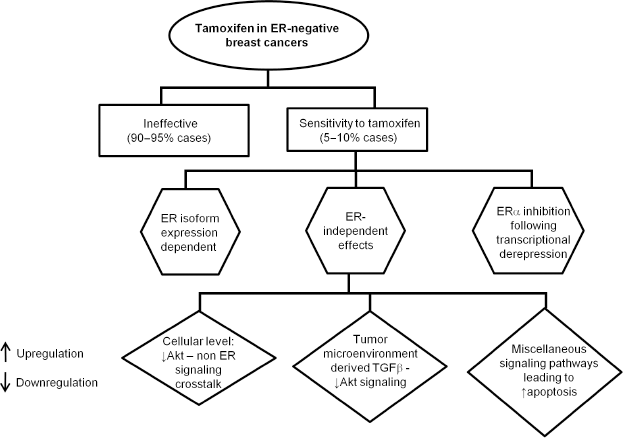
A schematic representation of known mechanisms of tamoxifen action in ER-negative breast cancer cells. The upward and downward arrows denote the upregulation and downregulation of any particular signaling component, respectively. The majority of ER-negative breast cancers (90-95% cases) are tamoxifen insensitive. In the remaining (5-10%) ER-negative breast cancer cases, tamoxifen combined with other agents decreases cell proliferation and induces apoptosis by different mechanisms.
ER-subtype-dependent Effects of Tamoxifen in ER-negative Breast Cancers
ER-negative breast cancer cells are not absolutely devoid of ER expression, although they are usually assigned ER-negative status based on the low expression status of the ERα isoform. Several studies investigating the effect of tamoxifen in ER-negative breast cancer cells with differential expression levels of ER isoforms have been performed.
ERβ influences tamoxifen response in ER-negative breast cancers
Breast cancers can be classified into several subgroups according to their molecular profile–-ER-positive subtypes luminal A (high ER expression) and luminal B (low ER expression), ER-negative HER-2-enriched subtype, and triple-negative breast cancer (TNBC) or basal subtype that is devoid of ER, PR, and HER-2 expression and is characterized by highly aggressive phenotype with poor prognosis.19–21 The majority of the breast cancer patients treated with tamoxifen are ER-positive type, and ERα serves as the predictive marker for assessing tamoxifen sensitivity. ER status is determined either by estradiol-binding assay detecting both ERα and ERβ or by immunoassays responding only to ERα.22–24 Interestingly, ~5-10% of the ERα-negative breast tumors have shown sensitivity to tamoxifen therapy.25–27 Studies assessing the role of ERβ are important because targeting this receptor subtype may bring more breast cancer patients under the purview of endocrine therapy. Several studies focus on the importance of ERβ expression as prognostic and/or predictive marker and on its role in regulating cancer cell proliferation and apoptosis. 28
According to recent studies, normal mouse and human mammary glands express more ERβ than ERα, and mammary epithelial cells of ERβ knockout mice are hyperproliferative indicating the tumor suppressor role of ERβ. 11 ERβ variation resulting in the difference in ERα/ERβ ratio has been associated with the modulation of response of breast cancer cells to hormone therapy. 29 ERβ has also been implicated in responsiveness of a fraction of ERα-negative breast cancer patients to tamoxifen. 28 Jensen et al in a study using tumor samples collected from 29 patients with invasive breast cancer showed that ERα-negative cells expressing ERβ express increased levels of the proliferation markers ki67 and cyclin A as compared to the ERα-positive breast cancer cells with the positive expression of ERβ and suggested an AP-1-dependent mechanism for tamoxifen insensitivity of these cells in the presence of ERβ. They have also indicated the benefit of using ERβ antagonists in combination with tamoxifen in reducing the proliferation in these cells. 12 In a study by Saal et al, ERβ protein levels have been found predictive for response to tamoxifen treatment in a large cohort of patients undergoing adjuvant tamoxifen therapy for two years, harboring both ERα-positive and ERα-negative tumors. There was a strong positive correlation between the fraction of the 353 patients expressing ERβ with ERα-negative status (55 patients) and distant disease-free survival. 30 Reese et al also showed that ERβ1-expressing MDA-MB-231 cells, a cellular model of TNBC, became sensitive to tamoxifen treatment upon stimulation by doxycycline in an ERβ1-mediated mechanism. Moreover, ER-negative subgroup of patients with the moderate expression of ERβ1 exhibited increased recurrence-free breast cancer survival (RFS) after adjuvant tamoxifen therapy. 31
In another study involving a cohort of early breast cancer premenopausal women, Yan et al showed better RFS of tamoxifen-treated ER-negative breast cancer patients expressing high levels of both ERβ1 and ER activity regulator steroid receptor RNA activator protein (SRAP) than those expressing either low ERβ1 or low SRAP as compared to the placebo-treated patients. 32 Based on the evidence from the abovementioned studies, ERβ expression status can be implicated as an important determinant for the sensitivity to tamoxifen treatment in ER-negative breast cancer patient population.
Effect of tamoxifen on GPER
Several research reports investigated the expression of membrane-bound G-protein-coupled estrogen receptor (mGPER30) in TNBC cells, which may stimulate the proliferation of these cells as a result of tamoxifen treatment.33–35 Girgert et al showed that the expression of mGPER30 in TNBC cell lines MDA-MB-435 and HCC-1806 caused increased cell proliferation upon stimulation by 4-hydroxytamoxifen (4-OHT), an active metabolite of tamoxifen. Knockdown of GPER30 prevented the proliferation of these cells by a mechanism that may involve the inhibition of Src kinase and epidermal growth factor (EGF) receptor by 4-OHT. 36
ER-independent Mechanisms and Effects of Tamoxifen
According to a few studies, tamoxifen appears to have both ER-dependent and ER-independent activities.37–39 In this context, the dose of tamoxifen used in the treatment may play an important role in determining whether tamoxifen will exert its usual ER-specific effect by acting as SERM when used in nanomolar dose range (100 nM to <1000 nM) in ER-positive cells or its ER-independent effects both in ER-positive and in ER-negative breast cancer cells when used in dose range as high as ≥1 μmol.40–42 Unlike its highly selective effect on ER in ER-positive breast cancer cells, tamoxifen has been found to exhibit off-target proapoptotic effects in ER-negative breast cancer cells.37,43–46 Some of the prominent ER-independent mechanisms reported by various studies are discussed in the following sections.
Akt phosphorylation mediates tamoxifen sensitivity in ER-negative MDA-MB-231 cells
Tamoxifen induces apoptosis in MDA-MB-231 cells by reducing Akt phosphorylation at Ser473 when used in combination with the PDK1 inhibitor OSU-03012 by suppressing Akt downstream effector proteins FOXO3a, GSK3a/b, and p27. The abovementioned combination also suppresses MDA-MB-231 tumor xenograft growth by 50% compared to 30% and 0%, respectively, when OSU-03012 and tamoxifen were used alone. Thus, combination with PDK1-mediated Akt inhibition sensitizes MDA-MB-231 cells to tamoxifen. 47
Protein phosphatase 2A (PP2A) is an important tumor suppressor protein in breast cancer, and cancerous inhibitor of PP2A (CIP2A) is overexpressed in many cancers, including breast cancer.48,49 Downregulation of CIP2A and phospho-Akt was correlated with tamoxifen-induced apoptosis in ER-negative breast cancer cells. Liu et al also showed that the overexpression of CIP2A or Akt reduced the tamoxifen-induced apoptotic effects in tamoxifen-sensitive breast cancer cells. On the contrary, knockdown of CIP2A by siRNA transfection sensitized tamoxifen-resistant cells to tamoxifen-induced apoptosis. Moreover, an in vivo study in nude mice showed that CIP2A downregulation inhibited xenograft growth. Overall, tamoxifen induced apoptosis in ER-negative cells in a dose- and time-dependent manner via the down-regulation of CIP2A/PP2A/p-Akt signaling, leading to tamoxifen-induced apoptosis. 50 Thus, the suppression of Akt phosphorylation combined with other ER-independent molecular targets can emerge as an effective therapeutic strategy to augment tamoxifen sensitivity in ER-negative breast cancer.
The important role of Akt signaling in determining tamoxifen sensitivity is not only restricted to the tumor cells but also evident in the interaction of ER-negative breast tumor cells and their microenvironment mediated by key signaling players, such as transforming growth factor beta (TGFP). These signaling cross talk events have been reviewed in the section focused on the tumor microenvironment later in this article.
Estrogen-related receptor alpha is a marker of tamoxifen sensitivity
Estrogen-related receptor alpha (ERRα) is an orphan nuclear receptor and an important component of signaling networks in breast cancer cells. 51 ERRα meets the metabolic demands of both normal cells and cancer cells requiring high energy for their specialized activities. In addition, the structural similarity of ERα and ERRα also makes it worthwhile to study the impact of ERRα expression on tamoxifen sensitivity in breast cancer patients without ER expression. Suzuki et al showed that high ERRα expression is associated with worse RFS and prognosis when neoadjuvant tamoxifen was given to 10% of the breast cancer patients of a small cohort (102) with ERα-positive status. 52 Because ERRα is expressed in ER-negative breast cancer, we have a unique opportunity to review how the modulation of its activity may affect the response to tamoxifen.27,53,54 In a recent study, our research group, using the microarray dataset from 2000 breast tumor patients from the medical centers worldwide and tumor samples derived from 910 breast cancer patients who participated in a tamoxifen-randomized primary breast cancer trial during 1976 through 1990 in Stockholm, Sweden, has established that ERRα expression predicts response to tamoxifen treatment. Moreover, we showed that ERRα could be a biomarker of tamoxifen sensitivity and a prognostic factor in TNBC. It will be important to confirm our results in other larger cohorts of patients with TNBC, randomized based on the expression of ERRα, and uniformly treated with tamoxifen. 55
ER coactivators such as AIB1 affect tamoxifen sensitivity
ER coactivator amplified in breast cancer 1 (AIB1) overexpression has been implicated in the development of both ER-positive and ER-negative breast cancers and tamoxifen resistance.56,57 Heck et al established a positive correlation between ERRα and AIB1 transcripts and associated resistance to tamoxifen therapy in tumor samples collected from 48 breast cancer patients from Heidelberg, Germany, during 2001 through 2003. 57 Weiner et al showed significant increase of RFS in ER-positive breast cancer patients with intermediate or high AIB1 expression when compared with ER-positive breast cancer patients with low AIB1 expression in response to adjuvant tamoxifen therapy, involving a cohort of 1780 post-menopausal women with breast cancer in Sweden during 1976 through 1990. In the same cohort, ER-negative subgroup with elevated AIB1 expression tended to show improved prognosis without adjuvant tamoxifen therapy. 58 Louie et al proposed that both ER-dependent signaling and ER-independent signaling mediated by AIB1 and E2F1, one of the key cell cycle regulators, are responsible for resistance to antiestrogens. They showed that ectopic expression of AIB1 induces the expression of key cell cycle regulators such as E2F1, E2F3, cyclin E, cyclin A, and Cdk2 that are different from those induced in response to estrogen (E2) treatment such as cyclin D1 in ER-positive breast cancer cells T-47D with reduced expression of ERα. Moreover, in these cells, E2 treatment led to the restoration of ERα expression. However, ectopic expression of AIB1 could not restore ERα expression, indicating that ERα-independent proliferation mediated by AIB1 is responsible for resistance to antiestrogens in ER-positive breast cancer cells. In addition, ER-independent association of AIB1 and E2F1 has also been shown as the causative factor for the proliferation of ER-negative breast cancer cells, such as HCC 1806, HCC1937, and BT-20, as seen by the upregulation of transcription of key cell cycle regulators. Thus, AIB1 may be a potential therapeutic target in ER-negative breast cancer cells that are resistant to tamoxifen therapy. 59
In addition, EGF receptor (EGFR) is a highly expressed protein in ER-negative breast cancer cells and is involved in regulating cell proliferation, apoptosis, angiogenesis, tumor invasiveness, and metastasis.60–62 Scandlyn et al showed that the combined effect of epigallocatechin gallate (EGCG) and tamoxifen significantly reduced tumor growth in female athymic nude mice implanted with MDA-MB-231 cells. They also established that this effect was primarily driven by the decline in the expression of the proteins EGFR, mechanistic target of rapamycin, and CYP1B1. 63 Previous studies also reported the role of tamoxifen-induced protein kinase C activation and subsequent activation of proapoptotic MAP kinase family member JNK1 by interfering with glycosphingolipid metabolism as mechanisms of apoptotic death of ER-negative breast cancer cells.28,64–66 Studies using c-MYC antisense oligonucleotide in ER-negative MDA-MB-231 cells observed the induction of apoptosis and also suggested the role of overexpressed c-MYC in the proliferation of cells treated with tamoxifen. 67
Tumor Microenvironment and TGFβ Signaling Play an Important Role in Determining Tamoxifen Response in ER-negative Breast Cancer
Tumor stroma is responsible for maintaining the growth of epithelial cells by secreting several growth factor and cytokines that regulate tumor growth, proliferation, invasiveness, and metastasis. The basement membrane acts as a separator of epithelial and endothelial cells from the stromal components. Stroma contains inflammatory cells and fibroblasts that make the proteins for extracellular matrix. 68 A study by Colletta et al showed that tumor fibroblasts in stroma secrete large amount of inhibitory growth factor TGFβ in response to tamoxifen despite the absence of ERs in these cells. This study suggests that tamoxifen can act in the absence of ER and the secretion of growth factor can help in the proliferation of tumor epithelial cells. 69 Interestingly, fibroblast-derived TGFβ in the tumor microenvironments of both ER-positive and ER-negative breast cancers has been reported to have a paracrine effect on each cell type growth according to several studies. Fibroblasts derived from ER-negative tumor patients have been shown to induce tamoxifen insensitivity in ER-positive breast cancer cells cocultured with those fibroblast cells as a result of increased levels of TGFβ secretion from the fibroblast cells. On the contrary, a study reported by Knabbe et al showed that TGFβ produced in the milieu of ER-positive MCF7 breast cancer cells can act in a negative paracrine manner to inhibit the growth of ER-negative breast cancer cells cocultured with ER-positive MCF7 breast cancer cells in response to tamoxifen treatment.40,53,70 Moreover, TGFβ suppresses cell proliferation and promotes cellular differentiation and apoptosis in normal breast tissues and the early stages of breast cancer, whereas the same TGFβ promotes cell migration, invasion, and metastasis by inducing epithelial-to-mesenchymal transformation (EMT) in the later stages of breast cancer. 71
TGFβ-Akt signaling and their mediators regulating tamoxifen action
Twist-related protein 1 (TWIST1) is expressed in several ER-negative breast cancer cell lines and is known to be involved in metastasis. Phosphorylation of TWIST1 can induce EMT in these cells via hyperactivated phosphatidyl-inosite-3-phosphate (PI3K)/Akt signaling. Two recent studies also suggested a cross talk between TGFβ and PI3K/Akt signaling pathways mediated by TWIST1 phosphorylation and degradation as a mechanism of tamoxifen action, leading to decreased invasiveness and metastasis in ER-negative cells. Using in vivo mouse model, Xue et al associated TWIST1 phosphorylation in ER-negative 4T1 breast tumor cells with lung metastasis and established that TWIST1 phosphorylation is mediated by upregulated TGFβ signaling in cooperation with hyperactivated PI3K/Akt to promote metastasis. Interestingly, tamoxifen, when used 10 times the dose used in ER-positive breast cancer, stimulates TWIST1 degradation, a proteasome-mediated pathway to significantly reduce the invasiveness and metastasis ability in these cells in both in vitro and in vivo models.72,73 Involvement of the transcription factor nuclear factor of activated T-cells (NFAT) has also been found as a causative factor in transforming tumor suppressive effect of TGFβ to its tumor promoting role by inducing EMT and c-MYC expression.74,75
Cross talk between TGFβ signaling and other pathways
TGFβ, EGF, and estrogen are some of the potent regulators of tumor cell proliferation, as evidenced in the study by Mints and Souchelnytskyi. They showed differential inhibitory effect on cell proliferation of ER-positive MCF7 cells and ER-negative MDA-MB-231 cells when these cells were cotreated with TGFβ1, EGF, and estrogen. Interestingly, tamoxifen enhanced the proliferation inhibitory effect of TGFβ1 combined with EGF and estrogen on MDA-MB-231 cells. On the contrary, TGFβ1 is not required for the growth inhibition of ER-positive MCF7 cells by EGF and estrogen combination. Moreover, the effect of tamoxifen treatment on tumor cell proliferation also correlated with decreased tumor size. This study also indicates the role of cross talking between different signaling network regulating tumor cell proliferation that can influence tamoxifen sensitivity in ER-negative breast cancer cells. 76 Thus, tamoxifen treatment can be beneficial in the restoration of tumor-suppressive role of TGFβ in ER-negative breast cancers.
Epigenetics and Tamoxifen Action
Deregulated epigenetic events, such as aberrant methylation of ER promoter and histone modification, have been associated with the lack of ER expression and tamoxifen insensitivity in ER-negative tumors.27,77–83 Several studies utilized histone deacetylase (HDAC) inhibitors as an effective strategy to circumvent aberrant HDAC activity leading to the restoration of ER expression and tamoxifen sensitivity in ER-negative cells.84–92 Sharma et al used demethylating agent 5-aza-dC and HDAC inhibitor trichostatin A to restore activated ER in TNBC cell line MDA-MB-231 that led to the restoration of ER as a target of tamoxifen treatment as a result of transcriptional repression by chromatin modification. 77 Two recent studies using mouse models also divulged mechanisms, such as phosphorylation of sphingosine analog FTY720 and selective inhibition of SIN3 corepressor as novel strategies associated with the correction of epigenetic deregulation in ER-negative and TNBC leading to the restoration of tamoxifen sensitivity.93,94 Overall, these studies showed the promise of combination therapy by including well-studied epigenetic modulators resulting in the sensitivity of ER-negative tumor cells to hormone therapy.
Concluding Remarks and Future Directions
In this review, we addressed different aspects of cellular tamoxifen treatment, adjuvant and neoadjuvant tamoxifen therapies in ER-negative breast cancer on the basis of receptor subtypes, ER-independent molecular mechanisms, the interaction between tumor cells and their milieu, and the epigenetic influence on tamoxifen sensitivity. Better understanding of these topics may provide valuable insights into study of tamoxifen therapy of hard-to-treat subgroups of ER-negative breast cancer patients, such as TNBC population. The lack of validated targets coupled with cytotoxicity associated with the conventional chemotherapy, poor prognosis, and its invasive nature make this subgroup of breast cancer difficult to treat. Identification of novel cellular therapeutic targets and combination therapies can be an effective approach to treat this breast cancer subtype.
Exploring novel markers for tamoxifen therapy and search of effective chemical agents for combination therapy will be immensely helpful for the treatment of this disease. As an example, ERRα is expressed in TNBC, offering a unique opportunity to study how modulation of its activity may affect the response to tamoxifen. In addition, the use of combination of tamoxifen with other novel therapeutic agents acting on specialized receptors, such as retinoid X receptors, can emerge as a promising treatment strategy, as evidenced by a recent clinical trial. 95 Further research is needed to investigate the cross talk between various signaling pathways determining tamoxifen sensitivity of ER-negative breast cancer patient population, including those suffering from TNBC.
Author Contributions
Conceived the concepts: SM and MKH. Analyzed the data: SM and MKH. Wrote the first draft of the manuscript: SM and MKH. Contributed to the writing of the manuscript: SM and MKH. Agree with manuscript results and conclusions: SM and MKH. Jointly developed the structure and arguments for the paper: SM and MKH. Made critical revisions and approved final version: SM and MKH. Both author reviewed and approved of the final manuscript.