
Abstract
Chaperone therapy is a newly developed molecular approach to lysosomal diseases, a group of human genetic diseases causing severe brain damage. We found two valienamine derivatives, N-octyl-4-epi-β-valienamine (NOEV) and N-octyl-β-valienamine (NOV), as promising therapeutic agents for human β-galactosidase deficiency disorders (mainly GM1-gangliosidosis) and β-glucosidase deficiency disorders (Gaucher disease), respectively. We briefly reviewed the historical background of research in carbasugar glycosidase inhibitors. Originally NOEV and NOV had been discovered as competitive inhibitors, and then their paradoxical bioactivities as chaperones were confirmed in cultured fibroblasts from patients with these disorders. Subsequently GM1-gangliosidosis model mice were developed and useful for experimental studies. Orally administered NOEV entered the brain through the blood-brain barrier, enhanced β-galactosidase activity, reduced substrate storage, and improved neurological deterioration clinically. Furthermore, we executed computational analysis for prediction of molecular interactions between β-galactosidase and NOEV. Some preliminary results of computational analysis of molecular interaction mechanism are presented in this article. NOV also showed the chaperone effect toward several β-glucosidase gene mutations in Gaucher disease. We hope chaperone therapy will become available for some patients with GM1-gangliosidosis, Gaucher disease, and potentially other lysosomal storage diseases with central nervous system involvement.
Keywords
Introduction
Lysosome is one of the cellular organelles where various high molecular endogenous or exogenous compounds are systematically digested under the acidic condition. 1 This physiological catalytic process is disturbed if mutations occur in one of the genes coding for the hydrolytic enzymes in the lysosome. Celllular dysfunctioin caused by an excessive storage of substrates ensues, and a genetic metabolic disease (lysosomal disease) develops clinically in humans and other animals with neurological and other somatic manifestations. This concept was first proposed for glycogen storage disease type I. 2
Since the mid-1960s, attempts have been made to the development of therapy for patients with lysosomal diseases. Theorectically enzyme replacement therapy was the most promising approach, and eventually shown to be effective for Gaucher disease patients, the most prevalent metabolic storage disorder of humans. 3 This approach has been extended to other lysosomal diseases. However, the effect has not been confirmed to brain pathology in patients with neurological manifestations.
GM1-gagliosidosis is one of the lysosomal diseases with storage of ganglioside GM1, keratan sulfate, and glycoprotein-derived oligosaccharides, presenting clinically with progressive neurological deterioration mainly in infancy and childhood. 4 This disease has been our major target of research for more than 40 years. We analyzed correlation of phenotypic manifestations with storage compounds, 5 enzyme activities,6,7 and enzyme molecules.8,9 Finally we moved to molecular pathology of β-galactosidase. 10
In parallel with with these experiments, in the early 1990s, we started molecular analyses of two genetically distinct human disease groups, β-galactosidosis (β-galactosidase deficiency disorders) caused by β-galactosidase gene mutations 4 and Fabry disease caused by α-galactosidase A gene mutations. 11 A paradoxical phenomenon was found that galactose itself and anologous low molecular weight competitive inhibitors could serve as chemical chaperones to induce expression of catalytic activities of mutant enzymes after stabilization and successful intracellular transport to the lysosome in the cells. We reported this enhancement first in Fabry disease,12,13 and then in GM1-gangliosidosis 14 and Gaucher disease. 15
After our early studies on galactose and 1-deoxygalactonojirimycin (DGJ), we developed new valienamine derivatives, N-octyl-4-epi-β-valienamine (NOEV) and N-octyl-β-valienamine (NOV) as chemical chaperones for mutant β-galactosidase and β-glucosidase proteins, respectively, to restore the enzyme activity in somatic cells from patients with GM1-gangliosidosis and Gaucher disease.14–16 We hope that this phenomenon will be applied to development of novel molecular therapeutic approach to lysosomal diseases, particularly with severe brain damage, in the near future. In this article we summarize our experimental results of chaperone effect and chaperone therapy mainly on NOEV for GM1-gangliosidosis.
Competitive Inhibitors of Lysosomal Enzymes
Carbasugar Glycosidase Inhibitors and Related Bioactive compounds
Carbasugars, previously known as pseudosugars, are a family of sugar mimics currently attracting interest among researchers in glycobiology and chemistry fields.17–19 The first example of carba-α-talopyranose was synthesized and called “pseudosugars.”
20
Later, naturally occurring bioactive carbasugar, 5a-carba-α-D-galactopyranose, was discovered.
21
Carbasugars are (hydroxymethyl)-branched-chain cyclitols. They are topologically similar to normal sugars particularly in the arrangement of the hydroxyl and hydroxymethyl groups, but have the oxygen atom of the pyranose or furanose ring replaced by methylene. Humans cannot differentiate carbaglucose from true glucose by their taste.
22
Furthermore carbahexopyranoses exist in structurally stable α- and β-anomer forms which are not interconvertible. Therefore, chemical modification at C-1 positions may be possible, providing biologically interesting compounds. Potent glycosidase inhibitors NOEV
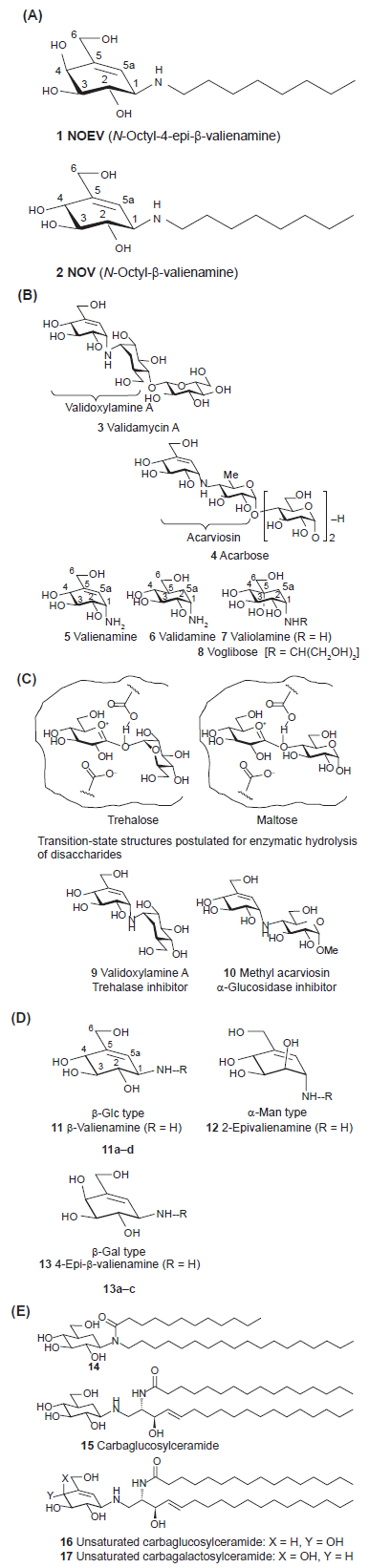
In 1970, agrochemical antibiotic validamycin A
Structural Modification of Valienamine
The enzyme inhibitory potency of
One of the authors (SO) became interested in chemical modification of
Glycocerebrosidase Inhibitors: Carbaglycosylceramides
Some glycosylamides are significant immunomodulators.
32
We prepared some carbasugar analogues, such as compound
Then a carbasugar mimic
N-Alkyl Valienamines, Potent β-Galactosidase Inhibitors
Complex ceramide chains were synthetically inaccessible. We therefore began to modify the structure by introducing a simple substitution of ceramide chain. Replacement with a simple aliphatic chain resulted in an increase of inhibitory activity
36
(Fig. 1D). Table 1 shows enzyme inhibitory activity of N-alkyl derivatives
Inhibitory activity [K i (µM)] of some N-alkyl-4-epi-β-valienamines against three glycosidases.
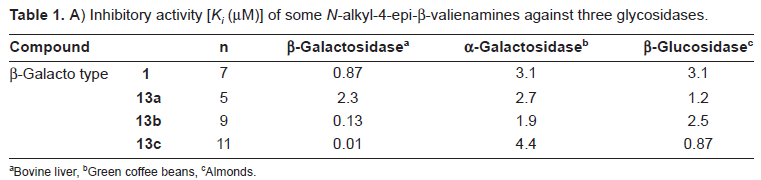
Bovine liver
Green coffee beans
Almonds.
Inhibitory activity [Ki (µM)] of some N-alkyl-β-valienamines against glucocerebrosidase.
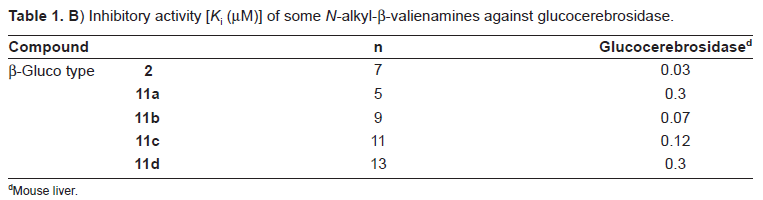
Mouse liver.
Preparative Studies on Valienamine Type Inhibitors
For further development of carbasugar chemistry, simple synthetic precursors are required. Diels-Alder endo-adduct
Typical routes to valienamines related to NOEV
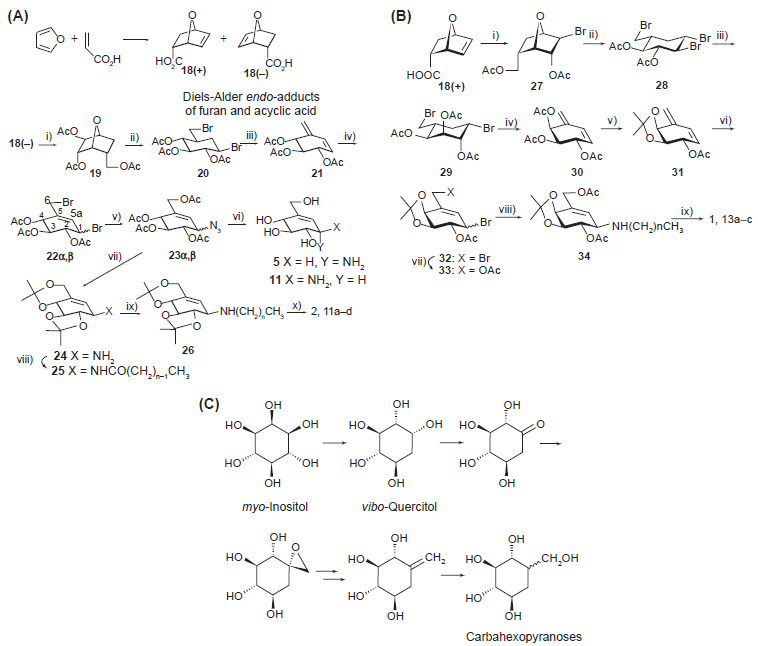
Effective N-alkylation of
NOEV
Transition-state type glycosidase inhibitor valienamines have thus been recognized as desirable carbohydrate mimics for designing new glycosidase inhibitors. Current technical difficulty is comparative inaccessibility to the carbahexose skeletons. Recently, we identified some routes to carbasugars through chemical transformation of (–)-vibo-quercitol derived from myo-inositol by biogenesis (Fig. 2C).42,43 This route establishes a link between naturally abundant cyclitols and chiral carbasugars.
Future Aspect
Carbaglycosylamines are chemically stable (hydroxymethyl) aminocylitols and expected to play roles as non-hydrolyzable mimics of glycopyranosylamines.43,44 Diverse modification of the biochemical and topologic nature of carbaglycosylamines may be achieved by substitution at the anomeric position, unsaturation at C-5 and C-5a, and hydroxylation at C-5 and/or C-5a, leading to improvement of biological function. As shown by the inhibitory activity of the N-alkyl derivatives
β-Galactosidosis: Genetic Human Disorders Caused by β-Galactosidase Gene Mutations
Lysosomal β-galactosidase (EC 3.2.1.23), encoded by a gene GLB1 (3p21.33), catalyzes hydrolysis of ganglioside GM1 and related glycoconjugates such as oligosaccharides derived from glycoproteins and keratin sulfate in human somatic cells. Allelic mutations of the gene result in excessive storage of the substrates in various cells and tissues.
GM1-gangliosidosis (OMIM 230500) is expressed clinically as generalized neurosomatic disease in children (infantile form, juvenile form), and rarely in adults (adult form), caused by widespread abnormal storage of ganglioside GM1, mucopolysaccharide keratin sulfate and glycoprotein-derived oligosaccharides in the central nervous system, skeletal system, and other tissues and visceral organs. Specific gene mutations are known for each clinical form. 45 Morquio B disease (OMIM 253010) is another clinical phenotype presenting with generalized skeletal dysplasia without neurological involvement. Again specific gene mutations different from those in GM1-gangliosidosis have been identified. 46 More than 100 gene mutations are collected, and successful gene diagnosis is well established using restriction enzymes specific to individual mutations. 4
At present only symptomatic therapy is available for the brain lesion in human GM1-gangliosidosis patients. Enzyme replacement therapy is currently in use for clinical practice for Gaucher disease, Fabry disease and other lysosomal diseases. However, the beneficial effect has not been confirmed for the brain damage, although general somatic signs and symptoms are clearly improved by continuous enzyme replacement therapy. 47 Secretion of feline β-galactosidase was reported in the transfected cell culture system, but the effect on the central nervous system was not shown. 48
After several years of basic investigations mainly for mutant α-galactosidase A in Fabry disease, we proposed chemical chaperone therapy for brain pathology in GM1-gangliosidosis, using an in vitro enzyme inhibitor N-octyl-4-epi-β-valienamine (NOEV) (1; Fig. 1A), a chemical compound newly produced by organic synthesis described above, 38 as a potent stabilizer of mutant β-galactosidase in somatic cells from patients with this disorder. 14
Genetic Metabolic Diseases: Molecular Pathology and an Approach to Possible Molecular Therapy
Molecular pathology of inherited metabolic diseases can be classified into the following three major conditions. 49
Biosynthetic defect of the protein in question. Mutant enzyme is not synthesized, and accordingly rescue of the protein is not possible.
Defect of biological activity. In spite of normal biosynthesis, the protein does not maintain biological activity because of its structural abnormality. There is no possibility to restore the biological activity of this molecule.
Unstable mutant protein with normal or near-normal biological activity under appropriate environmental conditions. The mutant protein has normal biological function in its mature form. However, it is unstable and rapidly degraded immediately after biosynthesis. The protein function is expected to be restored if the molecule is somehow stabilized and transported to the cellular compartment where it is expected to exhibit biological activity; the lysosome in the case of lysosomal enzyme.
We tested these possibilities first in Fabry disease, and found some mutant enzyme proteins were unstable at neutral pH in the endoplasmic reticulum/ Golgi apparatus, and rapidly degraded because of inappropriate molecular folding. 50 Addition of galactose in the culture medium of lymphoblasts from Fabry patients and COS-1 cells expressing mutant enzyme proteins surprisingly induced a high expression of α-galactosidase A activity. 12 However, a high concentration of galactose was necessary for treatment of these enzyme-deficient cells in culture. We concluded that a long-term treatment with galactose at this high dose was not realistic, although a short-term human experiment was reported on the beneficial cardiac function in a case of Fabry disease. 51 Accordingly we searched for other compounds that could enhance the enzyme activity in mutant cells. As stated above, DGJ was found to be effective for stabilization and high expression of the enzyme activity.13,52 DGJ showed the chaperone effect mainly on mutant α-galactosidae A. Its activity toward mutant β-galactosidase was 50-fold lower in a culture system experiment. 53
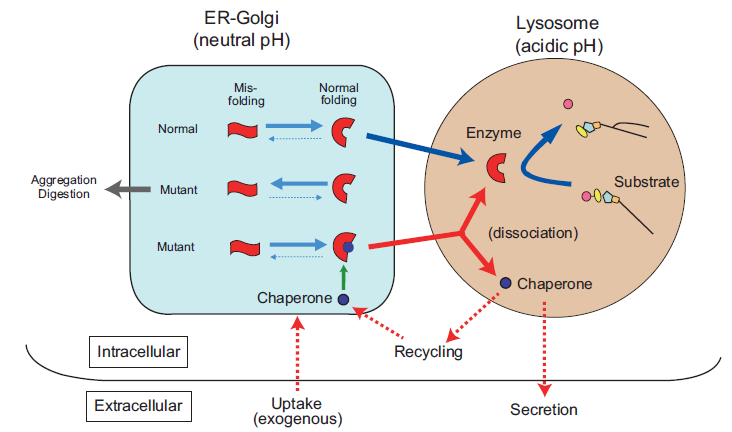
After subsequent extensive molecular analysis we reached the following conclusion. 49 A substrate analogue inhibitor binds to the misfolded mutant lysosomal protein as a kind of molecular chaperone (chemical chaperone), to achieve normal molecular folding at the endoplasmic reticulum/Golgi compartment in somatic cells, resulting in formation of a stable molecular complex at neutral pH. The protein-chaperone complex is safely transported to the lysosome, where it dissociates under the acidic conditions, the mutant enzyme remains stabilized in its normal folding, and its catalytic function is expressed (see below; Fig. 4).
Therapeutic Approach to Brain Pathology by Chemical Chaperones
New Chaperones: Valienamine Derivatives
We had particular interest in primary neuronopathic lysosomal diseases. Accordingly after studies on galactose and DGJ for α-galactosidase A, we started an extensive search for specific compounds for β-galactosidase. No commercially available compound was found as bioactive chaperone. Fortunately we came across two synthetic valienamine derivatives: β-galactosidase inhibitor N-octyl-4-epi-β-valienamine (NOEV) and β-glucosidase inhibitor N-octyl-β-valienamine (NOV) (
NOEV is a potent inhibitor of lysosomal β-galactosidase in vitro. It is stable and soluble in methanol or DMSO. The hydrochloride salt is freely soluble in water. Molecular weight is 287.40. IC50 is 0.125 µM toward human β-galactosidase. 14
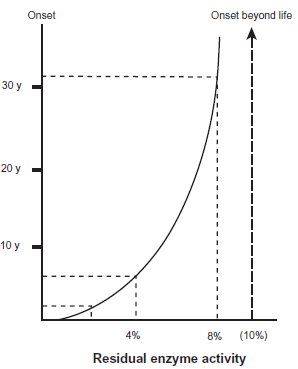
NOEV is 50-fold more active than DGJ in chaperone effect on mutant human β-galactosidase in GM1-gangliosidosis. Our calculations suggest that at least 10% of normal enzyme activity is necessary for washout of the storage substrate in lysosomal diseases (Fig. 3). The age of onset in patients expressing enzyme activity above this level is theoretically beyond the human life span. The same calculation was reported on some other lysosomal diseases. 54 We anticipate that the effective NOEV concentrations in human cells and animal tissues are much lower than the IC50 calculated in vitro, based on the results of tissue concentration after oral NOEV administration in experimental model mice. 55 In fact, NOEV is effective at the IC50 concentration in the culture medium for enhancement of mutant enzyme activity. 16 We hope it will be clinically used as a specific enzyme enhancer without exerting inhibitory effect in the cells.
Chaperone Experiments on Culture Cells
About one-third of the cultured fibroblast strains from GM1-gangliosidosis patients responded to NOEV; mainly from juvenile form and some infantile form patients. The effect is mutation-specific. 16 The R457Q mutant responded to NOEV maximally at 0.2 µM, and the R201C or R201H mutant at 2 µM. The mouse fibroblasts expressing mutant human β-galactosidase showed essentially the same results. 53 Molecular interaction between the chaperone and mutant protein depends on the structural modification of the mutant enzyme protein (Sakakibara et al. unpublished data). Addition of ganglioside mixture in the culture medium increased intracellular GM1 in the R201C cells causing juvenile GM1-gangliosidosis. 14 This storage was almost completely prevented by NOEV.
Chaperone Experiments on Genetically Engineered GM1-Gangliosidosis Model Mice
For animal studies we developed a knockout (KO) mouse strain with complete deficiency of β-galactosidase, 56 and then a transgenic (Tg) strain based on KO, expressing human R201C mutation (4% of the enzyme activity in the brain from wild-type mice). 14 Both showed neurological deterioration with some difference in severity. Life span was 7–10 months for KO and 12–18 months for Tg. Neuropathology corresponded to the clinical severity. 14 Short-term oral NOEV administration resulted in significant enhancement of the enzyme activity in all the R201C mouse tissues examined, including the brain. 14 Immunohistochemstry revealed an increase in β-galactosidase activity and decrease in GM1 and GA1 storage.
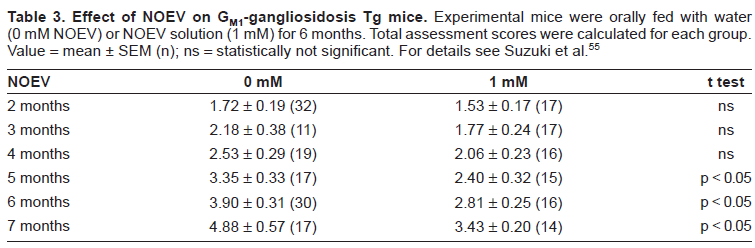
Oral NOEV treatment for the R201C Tg mouse showed an increase of the NOEV content in the brain in parallel with β-galactosidase activity, and GM1 storage decreased. 55 NOEV disappeared rapidly, within a few days after withdrawal. In this study we tried a new scoring system for neurological assessment 57 (Table 2). Treatment at the very early clinical stage (2 months) resulted in a positive clinical effect within a few months, although complete arrest or prevention of disease progression was not achieved under this experimental condition (Table 3). The latency before the clinical effect was longer if the therapy was started in the late symptomatic stage (6 months). We concluded that NOEV treatment at the early stage of disease is mandatory for prevention of the brain damage.

This result indicated the following sequence of events in the mouse brain. 49 After oral administration, NOEV goes directly into the bloodstream without intestinal digestion, is delivered to the brain through the blood-brain barrier, and restore the mutant β-galactosidase activity, resulting in substrate digestion and clinical improvement. No specific adverse effects have been observed for at least 6 months of continuous oral administration. For achievement of clinical drug development, however, we need to study further possible adverse effects and to establish the optinal dose and frequency of administration in order to achieve the best clinical effect.
Molecular Mechanisms of Chaperone Effect in Lysosomal Disease
As described above, β-galactosidase gene mutations result in excessive accumulation of substrates and various clinical phenotypes: GM1-gangliosidosis and Morquio B disease. Single base substitutions do not necessarily lead to a complete loss of enzyme function. However, the enzyme activity is not always expressed even if the potential catalytic function is not completely lost, simply because of intracellular instability of the mutant enzyme molecule due to inappropriate or incorrect protein folding. Molecular pathology of this type occurs at least in one-third of the patients with β-galactosidase deficiency. 16 Chemical chaperone corrects the molecular abnormality of this type, and assists intracellular transport to the lysosome, finally releasing the mutant enzyme as a stable bioactive protein (Fig. 4).
We postulated that enzyme-chaperone binding would become less strong under the acidic condition in the lysosome. Then the mutant enzyme molecule is released and stable catalytic activity appears. However, the precise mechanism of this NOEV effect is unknown at present. We therefore started computational analysis for prediction of molecular interactions between the β-galactosidase protein and the chaperone compound NOEV.
First, the three-dimensional structure of human enzyme was predicted employing a homology modeling method 3D-JURY,58,59 because the structure of this enzyme is not yet available. Penicillium sp. β-galactosidase was used as the template structure for homology modeling, and the predicted structure of human β-galactosidase has been obtained as shown in Figure 5A.
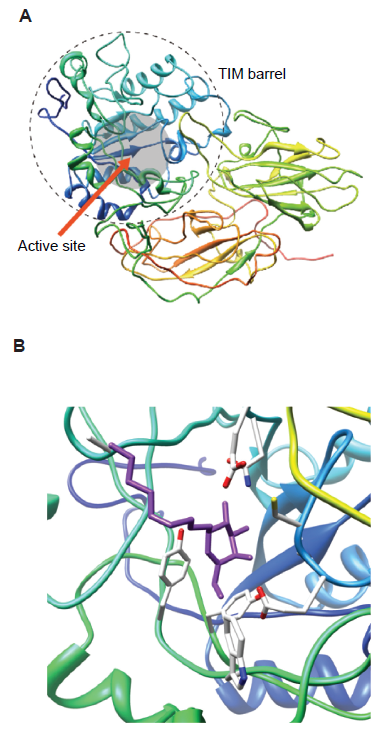
Second, plausible conformation of β-galactosidase-NOEV complex was determined in support of AUTODOCK4. 60 The conformation was subjected to further structural optimization. The result of the complex structure was successfully computed by AUTODOCK4 (Fig. 5B).
Third, the binding free energy of the two molecules in the complex was calculated by using AMBER9. 61 The computed binding free energy was −20.08 (kcal/mol) at pH 7.
Fourth, we calculated the effect of low pH in the lysosome on the binding affinity between the β-galactosidase and NOEV molecules. The low pH effect was represented as protonation of charged residues estimated by PROPKA. 62 The computed binding free energy at pH 5 was −18.06 (kcal/mol); higher than that at pH 7. This result indicates that affinity between β-galactosidase and NOEV is weakened at pH 5 compared with that at pH 7. Consequently, we concluded that (1) the enzyme-NOEV complex has lower free energy than the unbound enzyme, and (2) protonation of an active site residue causes free energy change consistent with the chemical chaperone hypothesis.
Conclusion
This new therapeutic strategy (chaperone therapy) is in principle applicable to all lysosomal diseases, if a specific compound is developed for each enzyme in question. We have already confirmed the effect in Fabry disease, GM1-gangliosidosis, and Gaucher disease. Other related diseases also are currently studied by other investigators.63,64 Theoretically this principle can be applied to all other lysosomal diseases. Furthermore, there may well be other genetic diseases to be considered, if molecular pathology in somatic cells has been clarified in detail. We hope studies in this direction will disclose a new aspect of molecular therapy for inherited metabolic diseases with central nervous system involvement in the near future.
Abbreviations
NOEV, N-octyl-4-epi-β-valienamine; NOV, N-octyl-β-valienamine; DGN, 1-deoxygalactonojirimycin; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene; LAH, lithium aluminum hydride; KO, knockout; Tg, transgenic.
Disclosure
The authors report no conflicts of interest.
Footnotes
Acknowledgments
This research was supported by grants from the Ministry of Education, Culture, Science, Sports, and Technology of Japan (13680918, 14207106), and the Ministry of Health, Labour and Welfare of Japan (H10-No-006, H14-Kokoro-017, H17-Kokoro-019). We thank all collaborators contributing to this research project for the past 16 years.