
Abstract
Anaerobic intestinal spirochaetes of the genus Brachyspira include both pathogenic and commensal species. The two best-studied members are the pathogenic species B. hyodysenteriae (the aetiological agent of swine dysentery) and B. pilosicoli (a cause of intestinal spirochaetosis in humans and other species). Analysis of near-complete genome sequences of these two species identified a highly conserved 26 kilobase (kb) region that was shared, against a background of otherwise very little sequence conservation between the two species. PCR amplification was used to identify sets of contiguous genes from this region in the related Brachyspira species B. intermedia, B. innocens, B. murdochii, B. alvinipulli, and B. aalborgi, and demonstrated the presence of at least part of this region in species from throughout the genus. Comparative genomic analysis with other sequenced bacterial species revealed that none of the completely sequenced spirochaete species from different genera contained this conserved cluster of coding sequences. In contrast, Enterococcus faecalis and Escherichia coli contained high gene cluster conservation across the 26 kb region, against an expected background of little sequence conservation between these phylogenetically distinct species. The conserved region in B. hyodysenteriae contained five genes predicted to be associated with amino acid transport and metabolism, four with energy production and conversion, two with nucleotide transport and metabolism, one with ion transport and metabolism, and four with poorly characterised or uncertain function, including an ankyrin repeat unit at the 5’ end. The most likely explanation for the presence of this 26 kb region in the Brachyspira species and in two unrelated enteric bacterial species is that the region has been involved in horizontal gene transfer.
Artemis Comparison Tool; base pair; clusters of orthologous groups; deoxyribonucleic acid; guanine plus cytosine; guanine plus cytosine at the third base position on a codon; horizontal gene transfer; kilobase; megabase; polymerase chain reaction.
The GenBank accession numbers for the genome region sequences for Brachyspira hyodysenteriae WA-1 and Brachyspira pilosicoli 95/1000 are EF694538 and EF694539, respectively.
Introduction
The genus Brachyspira currently includes seven officially named species of anaerobic Gram-negative spirochaetes that colonise the large intestine of various species of animals and birds (Hampson and Stanton, 1997). The best studied of these are the two pathogenic species B. hyodysenteriae and B. pilosicoli. B. hyodysenteriae is the causative agent of swine dysentery, an important disease of swine that is characterised by severe mucohaemorrhagic diarrhoea (Hampson et al. 2006). B. pilosicoli is the causative agent of intestinal spirochaetosis, a condition associated with mild colitis and diarrhoea. It is best known as a disease of pigs, but can also affect poultry, dogs, horses and human beings (Hampson and Duhamel, 2006).
Recent insights into Brachyspira species evolution and interactions have been obtained from investigations into gene transfer associated with a novel prophage-like agent (Stanton et al. 2001; Stanton et al. 2003; Matson et al. 2005), analysis of the population structures and species relatedness using multilocus enzyme electrophoresis (Trott et al. 1997; Trott et al. 1998) and multilocus sequence typing (Råsbäck et al. 2007), and investigations of genomic rearrangements between B. hyodysenteriae and B. pilosicoli by comparing limited physical genomic maps (Zuerner et al. 2004). The close similarities of the 16S rRNA sequences of the various Brachyspira species indicate that Brachyspira speciation has occurred more recently than it has in many other bacterial genera (Paster and Dewhirst, 1997). The sizes of the B. hyodysenteriae and B. pilosicoli genomes have been estimated at 3.2 megabase (Mb) and 2.45 Mb, respectively (Van Der Zeijst and ter Huurne, 1997).
In the current study, comparative analysis of near-complete genome sequences of B. hyodysenteriae and B. pilosicoli identified a highly conserved 26 kilobase (kb) genomic region shared by the two species, against a backdrop of otherwise low genome sequence conservation. This genomic region was not found in spirochaetes from other genera, but was partially conserved in Enterococcus faecalis and Escherichia coli.
Methods
Spirochaete strains
The Brachyspira species strains used in the study were obtained from the culture collection held at the Reference Centre for Intestinal Spirochaetes, Murdoch University, Western Australia. These included five strains each of B. hyodysenteriae (strains WA1, B204, P18A, B169, and SA3), B. pilosicoli (strains 95/1000, CSP-1, WesB, HRM7B, and P43/6/78T), four strains of B. intermedia (strains HB60, PWS/AT, P280/1, and 889), B. innocens (strains B256T, 4/71, 89/840, and UNL-2), and B. murdochii (strains UNL-1, 89/2209, 155.2, and 56/150T), three strains of B. aalborgi (strains 513T, W1, and W2) and two strains of B. alvinipulli (strains C1T and C2). The strains originated from different host species (pigs, chickens and humans), and were isolated in Australia, Europe, Scandinavia and North America. The two strains that were sequenced were isolated in Australia from pigs with diarrhoea. The identity of all strains used previously had been established using phenotypic testing, species-specific PCR assays (where available) and analysis of 16S rRNA gene sequences.
Media and culture conditions
Individual spirochaete strains were propagated at 37 °C in Kunkle's pre-reduced anaerobic broth containing 2% (vol/vol) foetal bovine serum and a 1% (vol/vol) ethanolic cholesterol solution (Kunkle et al. 1986). Cells were harvested from mid-log phase culture and counted using a haemocytometer chamber. For polymerase chain reaction (PCR) analysis, the harvested cells were resuspended with 10 mM Tris-HCl (pH 8.0) to a cell density of 104 cells/ml and lysed by boiling for 30 sec. Two μl of the resuspended cells was used directly for the assays.
Genomic sequencing and assembly
A whole genome shotgun sequence strategy was used to sequence B. hyodysenteriae strain WA-1 and B. pilosicoli strain 95/1000 under a commercial contract with the Australian Genome Research Facility, University of Queensland, Brisbane. Approximately 5x coverage raw sequence reads were assembled into contigs using Phred and Phrap (http://bozeman.mbt.washington.edu/phredphrap-consed.html), using default parameters.
Gene prediction
Gene predictions for both genomes were performed using Glimmer [version 2.13] (Delcher et al. 1999). To assign a putative function, each coding sequence was translated using the EMBOSS package (Rice et al. 2000) program SIXPACK, and then compared to the non-redundant protein database from NCBI using the BLASTp algorithm (BLAST version 2.2.6; Altschul et al. 1990). Matches were considered significant if there was greater than 30% sequence identity and 60% hit coverage (Konstantinidis and Tiedje, 2005). Signal peptide motifs and protein localisation were predicted for all translated coding sequences, using SignalP (Nielsen et al. 1997), and PSORT-B (Gardy et al. 2003), respectively.
Sequence composition
The guanine plus cytosine (GC) composition was calculated for all genes using the EMBOSS program GEECEE for overall GC-content, and EMBOSS program WOBBLE for GC-content at the third base of each codon (GC3).
Sequence comparisons
The assembled partial (~90%) genome sequences of the two spirochaetes were compared at the nucleotide level, by concatenating all contigs for each genome into “pseudo-genomes”, and then using the program Dotter version 3.1 (Sonnhammer and Durbin, 1995). Dotter was also used to compare the 26 kb region in B. hyodysenteriae WA-1 with a similar region in Enterococcus faecalis strain V583.
Similar region identification
The 26 kb region in B. hyodysenteriae WA-1 was compared to all available bacterial genomes from GenBank (548 bacterial genomes, as of 23/10/07) using the BLASTx algorithm from BLAST (Altschul et al. 1990), at a maximum e-value stringency of 10-30. Using Son of Eric (version 3.2; http://ccg.murdoch.edu.au/soe/), regions with clustered protein sequence matches were manually identified and selected for further investigation. In order to reduce redundancy (due to the high similarity between the E. coli strains), some analyses only included one strain of E. coli (E. coli K-12) as a species representative.
Similar region analysis
The similar regions were isolated from the genome sequence using EMBOSS program EXTRACTSEQ. Sequences were aligned using CLUSTALW (version 1.83.1, Thompson et al. 1994). Statistics of the alignments were obtained using the EMBOSS program INFOALIGN. The Percent Change was calculated as [(Align Length–Identical) × 100] divided by Align Length. The Percent Change value was the indicator for sequence similarity, with Percent Change inversely proportional to sequence similarity. The final nucleotide similarity values presented throughout this investigation were calculated by subtracting Percent Change from 100%.
The 26 kb region of B. hyodysenteriae WA-1, B. pilosicoli 95/1000, and E. faecalis V583 were compared using the tBLASTx algorithm from BLAST (Altschul et al. 1990). Results, with matches having a BLAST score greater than 400, were visualised using the Artemis Comparison Tool (ACT) (version 4; Carver et al. 2005). ACT was also used to predict coding sequence and GC composition of the similar region.
Gene order of the 26 kb region in B. hyodysenteriae WA-1, B. pilosicoli 95/1000, E. faecalis V583, Photobacterium profundum strain SS9, and E. coli strains Sakai (serotype O157:H7), EDL933 (serotype O157:H7), K-12, and CFT073 was investigated using the BLASTp algorithm from BLAST (Altschul et al. 1990). All proteins in the regions were compared, and significant matches were considered those with more than 30% identity covering more than 60% of the protein, and a maximum e-value of 9 × 10–30.
Gene function was putatively assigned from a Hidden Markov Model (HMM) built using the clusters of orthologous groups (COG) database (http://cogplus.tau.ac.il/; Tatusov et al. 2001) and using the program HMMer (Eddy, 1998). Function was assigned to coding sequences with a significant match (e-value less than 10–04) at the amino acid level.
Operon predictions for E. faecalis V583, E. coli strains Sakai, EDL933, K-12 and CFT073, and P. profundum SS9, were obtained from the VIMSS database (from http://www.microbesonline.org/operons/; Price et al. 2005).
Primer design and PCR for core genes
Nine primer pairs were designed, using Oligo Explorer version 1.1.0 (OligoSoftware), to investigate the contiguity of nine core genes (genes BH3-BH11 in B. hyodysenteriae WA-1) amongst the listed strains of the different Brachyspira species. Primer pairs were designed so that the resulting amplicons overlapped adjacent genes (Fig. 1). The primer sequences are listed in Table 1. Spirochaetal deoxyribonucleic acid (DNA) was amplified by long-range PCR in a 25 μl total volume using the Advantage™ 2 PCR Enzyme System (BD Biosciences), according to the manufacturer's instructions. Briefly, amplification mixtures consisted of 1 × BD Advantage Polymerase Mix, 0.2 mM of each deoxyribonucleotide triphosphate (Promega), 0.5 μM of forward primer, 0.5 μM of reverse primer, and 2 μl spirochaete chromosomal DNA. Cycling conditions involved an initial 5 min template denaturation step at 95 °C, followed by 30 cycles of denaturation at 95 °C for 30 sec, annealing at 50 °C for 30 sec, and a primer extension at 68 °C for 3 min. The PCR products were subjected to electrophoresis in a 1% (w/v) agarose gel in 1 × TAE buffer (40 mM Tris-acetate, 1 mM ethylenediamine tetraacetic acid), stained with ethidium bromide and viewed over ultraviolet light. All PCR analyses were performed in triplicate. The apparent molecular mass of the amplification products was determined by comparison with a 1 kb DNA ladder (New England Biolabs).
Diagramatic representation of the annealing position and amplicons of the nine primer sets used for PCR over nine genes (BH3-BH11) in the 26 kb region of B. hyodysenteriae WA-1. The primer sequences are shown in Table 1 Oligonucleotide primers used in PCR reactions for contiguity analysis of nine core genes in Brachyspira species strains.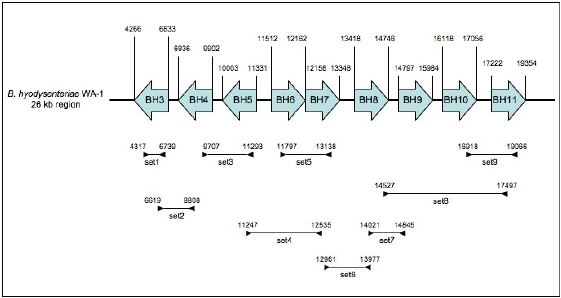
GenBank submission
The nucleotide sequences of the 26 kb regions in B. hyodysenteriae WA-1 and B. pilosicoli 95/1000 were submitted to GenBank, and were given accession numbers EF694538 and EF694539, respectively.
Results
Comparative genome analysis of the 26 kb region
A dot plot analysis conducted between the pseudo-genomes of B. hyodysenteriae WA-1 and B. pilosicoli 95/1000 at the nucleotide level revealed a single conserved 26 kb region (78% nucleotide similarity) amongst a background of low sequence similarity between the two species.
The coding sequences identified in the 26 kb region displayed significant similarity to sequences in 27 of the 548 published bacterial genomes that were analysed. Six genomes contained regions with gene clusters similar to the 26 kb region, these being E. faecalis V583 (base positions 2479008–2500903; 21.985 kb), P. profundum SS9 (base positions 2220950–2294950; 74 kb), and E. coli strains Sakai (base positions 3738676–3770965; 32.298 kb), EDL933 (base positions 3805982–3838273; 32.291 kb), K-12 (base positions 2998367–3022208; 23.841 kb), and CFT073 (base positions 3296747–3329078; 32.331 kb). None of the other available spirochaete genomes (from the genera Treponema, Borrelia and Leptospira) contained a similar gene cluster.
Sequence similarity and dot plot analyses of the B. hyodysenteriae WA-1 26 kb region and the similar region in E. faecalis V583 displayed the highest inter-genera nucleotide conservation (43.3% nucleotide similarity), while analyses of the B. hyodysenteriae WA-1 26 kb region with E. coli and P. profundum, displayed dispersed regions of similarity. Nucleotide similarity ranged from 35.5% to 40.2% for E. coli, depending on the strain.
Coding sequence %GC composition analysis gave averages of 29, 29, 37.2, 42 and 51 for B. hyodysenteriae, B. pilosicoli, E. faecalis, P. profundum and E. coli K-12, respectively. The %GC for the E. faecalis genes EF2573, EF2575, EF2577–EF2579, and EF2581–EF2583 (Fig. 2) was below 40%, similar to the %GC of the 26 kb region coding sequences in B. hyodysenteriae and B. pilosicoli (~35%), even though their genome averages are approximately 27%. Finally, for the coding sequences analysed in this investigation, no variation was identified between the %GC and the %GC at the third base position (%GC3) (data not shown).
Gene order and function of the similar regions in B. hyodysenteriae WA-1, B. pilosicoli 95/1000, and E. faecalis V583, are illustrated diagrammatically. Genes shared between the regions are highlighted with connecting purple lines. The genes in these regions have been designated a number of COG functional categories (http://www.ncbi.nlm.nih.gov/COG/), each represented by a different colour. Genes associated with metabolism are coloured blue, those associated with information storage and processing are green, those associated with cellular processes are red, unclassified functional group genes are yellow and those genes that did not associate with any of the functional groups in the database are white. Two horizontal black lines indicate the region with the greatest gene order conservation. The diagram illustrates a conservation of gene order, and gene function between the similar regions of the Gram-positive bacterium E. faecalis V583 and the Gram-negative spirochaetes B. hyodysenteriae WA-1 and B. pilosicoli 95/1000.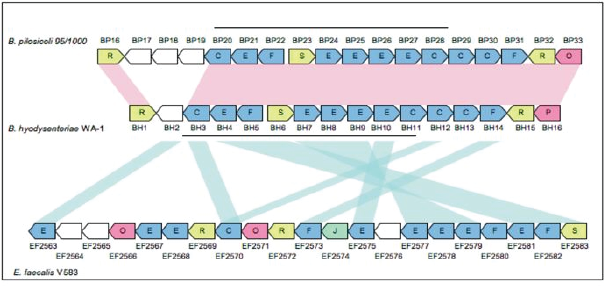
The base composition of the flanking genomic regions of the 26 kb region in B. hyodysenteriae, B. pilosicoli, E. faecalis, and E. coli K-12 (as representative of the E. coli strains), displayed distinct changes in GC content (%GC), where the average %GC in the flanking regions display an increase or decrease from the 26 kb region%GC. In B. hyodysenteriae (complete 26 kb region), B. pilosicoli (complete 26 kb region), and E. coli K-12 (a seven-gene cluster), the flanking regions displayed distinct changes of 15, 17, and 10 %GC respectively, while in E. faecalis (an eleven-gene cluster), the flanking regions displayed changes of 5 %GC. No distinct %GC changes were identified in the flanking regions of the two gene clusters in P. profundum.
The gene function prediction, gene organization, and gene similarity in the 26 kb region between the two Brachyspira species and E. faecalis are shown in Figure 2. Fifteen coding sequences were conserved as identical clusters between the 26 kb regions of B. hyodysenteriae and B. pilosicoli. Eleven coding sequences were conserved in E. faecalis, B. hyodysenteriae and E. coli strain CFT073, while 10 coding sequences were conserved in E. coli strains Sakai, EDL933 and K-12, and eight coding sequences were conserved in the P. profundum genome (data not shown).
Of the 16 genes in the B. hyodysenteriae 26 kb region, five were associated with amino acid transport and metabolism (COG functional group E), four were associated with energy production and conversion (COG functional group C), two were associated with nucleotide transport and metabolism (COG functional group F), one was associated with inorganic ion transport and metabolism (COG functional group P), and four, one of which was characterised as an ankyrin repeat protein (BH1), were either poorly characterised (COG functional groups R and S), or unknown (BH2, BH6, BH15). The gene order in the 26 kb region of B. hyodysenteriae WA-1 and B. pilosicoli 95/1000 was identical, except for a two-gene insertion in B. pilosicoli (Fig. 2). At the 5’ end of this 26 kb region, a distinct 2.5 kb ankyrin repeat sequence (COG0666 ankyrin repeat) was shared by the two species (genes BH11 and BP16, respectively). The ankyrin repeat sequence in B. hyodysenteriae and B. pilosicoli was absent from the similar regions in the other bacterial species. Other repeat sequences, predicted integrases and tRNA sites were not identified within 30 ORFs of the two Brachyspira 26 kb genomic regions (data not shown).
Gene clustering conservation between the B. hyodysenteriae WA-1 and E. faecalis V583 26 kb regions was found, with nine genes from a 14 gene cluster in E. faecalis found in B. hyodysenteriae (BH3–BH11) (Fig. 2). When compared to the order and orientation of the B. hyodysenteriae genes, the E. faecalis genes from EF2581 to EF2583 (BH4–BH6 in B. hyodysenteriae) showed the same order and direction, while genes EF2577 to EF2579 (BH7–BH9 in B. hyodysenteriae) were in reverse order and direction. In the E. coli strains and P. profundum, the genes similar to B. hyodysenteriae BH7, BH8 and BH9 were also arranged consecutively and in the same direction, but in different order (data not shown).
Operon predictions for the 26 kb similar regions in E. faecalis, P. profundum, and the E. coli strains indicate some likely operons exist within these conserved clusters. The most consistent operon prediction was in seven of the eight genomes (with the exception being P. profundum), where genes corresponding to BP24–BP26 in B. pilosicoli were predicted to be an operon.
Distribution of PCR products in Brachyspira species and strains
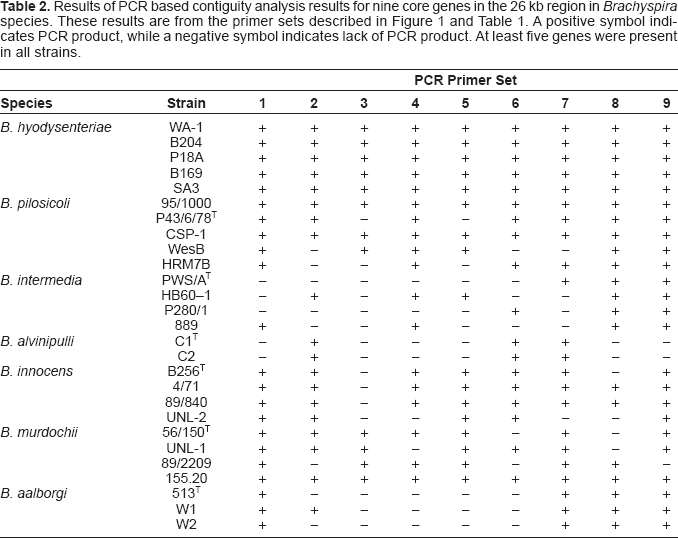
Results of PCR based contiguity analysis results for nine core genes in the 26 kb region in Brachyspira species. These results are from the primer sets described in Figure 1 and Table 1 A positive symbol indicates PCR product, while a negative symbol indicates lack of PCR product. At least five genes were present in all strains.
Discussion
Brachyspira hyodysenteriae and B. pilosicoli are both anaerobic spirochaetes that colonise the large intestine and initiate disease. Despite these similarities, the genomes of these two species lack genome sequence similarity. Nevertheless, against this background, the two species do share a 26 kb region that is highly conserved in nucleotide and amino acid sequence, and in predicted protein function.
Comparisons of the 26 kb region at both the nucleotide and amino acid sequence levels indicated that the coding sequences in this region are found in similar clusters in only two species, namely E. faecalis and the E. coli strains, while in P. profundum two small clusters were present. From an evolutionary perspective this similarity was unexpected, since the Spirochaetes are phylogenetically distinct from the Firmicutes and the Proteobacteria. Previously, similarities have been observed between the nicotinamide adenine dinucleotide oxidase genes in B. hyodysenteriae and E. faecalis, and in this case a common gene ancestry was suggested as an explanation (Stanton and Sellwood, 1999).
Three potential reasons for gene order conservation between species have been suggested (Tamames, 2001). The first involves the region being conserved through a common ancestor, in a process known as vertical transfer (Lawrence, 2005). If this were the case for the 26 kb region, it would be expected that it would be present in other Spirochaetes, and not in two taxonomically unrelated species. A second reason is that the integrity of the cluster is important to the fitness of the cell. Two features are associated with gene clustering and cell efficiency: these are transcriptional units, which are clusters of related genes for transcriptional and/or translational efficiency (Wolf et al. 2001), and operons, which are clusters of functionally related genes for regulation purposes (Rocha and Danchin, 2003; Pal and Hurst, 2004). A number of the genes present in the similar regions of B. hyodysenteriae and B. pilosicoli, and their similar proteins in E. faecalis, E. coli, and P. profundum, have been predicted to be associated with multiple operons. However, no known promoter regions or other regulatory elements were found in these regions and not all the clustered genes were directly involved in related cellular processes (data not shown). Hence it is unlikely that the whole region is a complete operon (Lathe et al. 2000). The third reason for gene order conservation is that it is a result of a horizontal gene transfer (HGT) event.
In support of the involvement of an HGT event as an explanation for the current observation, at the base composition level a distinctive sharp increase or decrease was observed in the GC content of the 26 kb region and in the similar regions in the other species. Such a pattern is characteristic of acquired DNA (Buchrieser et al. 2003), and in particular mobile genetic elements, such as pathogenicity islands (Schmidt and Hensel, 2004). Further evidence for a potential HGT event was the presence of an ankyrin repeat sequence at the 5’ end of the 26 kb region of both B. hyodysenteriae and B. pilosicoli. Repeat sequences are known to facilitate recombination, and also allow insertion of foreign DNA; for example, mobile genome islands (such as pathogenicity islands) may be flanked by direct repeats (Schmidt and Hensel, 2004). Furthermore, ankyrin repeats in bacterial genomes have been identified in close proximity to genes with functions that are commonly transferred, specifically genes associated with nutrient acquisition, and antibiotic tolerance or resistence (Nascimento et al. 2004).
Given the available evidence, the most likely explanation is that this 26 kb region is present in B. hyodysenteriae, B. pilosicoli, E. faecalis, and E. coli as a result of its involvement in HGT events. Where and when these event(s) occurred is uncertain, but the Brachyspira species, E. faecalis and E. coli are all enteric bacteria, and this shared environment may have provided a suitable venue for transfer to occur. The direction of the gene transfer is uncertain, and clearly other bacterial species that so far have not been sequenced could be the source of the original DNA that was involved in the transfer(s).
By using overlapping PCRs all the Brachyspira species investigated were found to contain five or more of the nine core genes from the 26 kb genomic region. The presence of at least some the core genes in the correct order in all the Brachyspira species, and their absence from other sequenced spirochaetes, suggests that if this region was acquired by Brachyspira species by a HGT event, it most likely occurred sometime after the foundation Brachyspira species became separated from other spirochaete genera, but before further Brachyspira speciation occurred. The apparent absence of some of the 26 kb region core genes in the different Brachyspira species may have been because the primers used were designed based on the B. hyodysenteriae sequence, and polymorphisms could be present within the primer sequences in the other species. Alternatively, the genes may have been absent or arranged in a different order, such that they were not amplified. Previously it has been deduced that gene rearrangements have been important in shaping the genomes of B. hyodysenteriae and B. pilosicoli (Zuerner et al. 2004), and similar gene rearrangements are likely to occur in other Brachyspira species. The presence or absence of these genes could be further investigated by using low stringency hybridisations.
In conclusion, a partially conserved 26 kb genomic region has been identified in Brachyspira spp., Enterococcus faecalis and Escherichia coli. Further work is required to investigate the origin and the functional significance of the 26 kb region in the various bacterial species in which it has been detected.
Footnotes
Acknowledgements
Yair Motro was supported by an Australian Research Council (ARC)—Industry linked scholarship in partnership with Novartis Animal Vaccines, who also funded the research. Thanks are due to Professor Trifinov at the Centre for Genome Diversity (University of Haifa) for helpful discussions.