
Abstract
The infectious agent of transmissible spongiform encephalopathy (TSE) was assumed to be the aggregate of abnormal prion protein isoform (PrPsc). We observed that lowering the pH of 3% SDS-inoculated plasma or brain homogenate after PK digestion to 4.5 (acidic SDS condition) enabled to precipitate proteinase K-resistant prion protein (PrPres) in plasma as well as PrPres in the brain with synthetic poly-A RNA as affinity aggregate. Therefore, we determined if RNA molecules could be used for discriminating TSE patients from healthy individuals. We also examined the plasma of patients with classical Creutzfeldt–Jakob disease (CJD) and other brain disorders who were not diagnosed with TSE. The results indicated that RNA approximately 1.5–2.0 kb in length was commonly observed in the plasma of patients with brain disorders but was not detected in the plasma of healthy volunteers. Enhanced expression of RNA and its protection from endogenous nucleases might occur in the former group of patients. Moreover, we speculate that the non-transmissible neuronal disorders overlap with prion diseases.
Introduction
Transmissible spongiform encephalopathies (TSEs) are fatal neurodegenerative diseases, which include sporadic Creutzfeldt–Jakob disease (spCJD), variant Creutzfeldt–Jakob disease (vCJD) and Gerstmann-Straüssler syndrome (GSS) in humans; bovine spongiform encephalopathy in cattle; scrapie in sheep and goats; and chronic wasting disease in deer. There is accumulating evidence that vCJD can be transmitted via transfusions of blood and blood components; these findings have raised concerns about the iatrogenic spread of this disease through blood among the general population.1–5 To address this etiological concern, it is necessary to develop presymptomatic diagnostic test methods using blood samples to identify vCJD-infected individuals from among the general population and discriminate them from other uninfected individuals.6,7
In addition, unlike the pre-symptomatic diagnostic blood tests for vCJD, there was no urgent need for the development of ante-mortem diagnostic tests for classical CJD, including spCJD, or other TSEs that seemed unlikely to be transmitted via transfusion of blood or blood products. However, development of an ante-mortem blood test to diagnose the so-called classical CJD and other TSEs could allow effective treatment and consequently improve patient survival; this development was as important as that of a pre-symptomatic blood test for diagnosing vCJD.8,9 The need for diagnostic tests was further emphasized by the results of the experiments on animal models which confirmed the secondary transmission of scrapie and classical CJD via blood transfusion. Therefore, the development of ante-mortem and presymptomatic blood tests for diagnosing TSEs, including vCJD, and the so-called classical CJD, should be given equal importance in TSE research. Such blood tests can facilitate the detection and analysis of disease outcome and lead to the development of methods for epidemiological and clinical research.
To date, ante-mortem diagnosis of TSEs was made using pathophysicochemical methods by employing various diagnostic machines or by using biopsied tissue preparations.10,11 Since TSEs are mainly characterized by degeneration of the brain, indirect methods using non-brain tissue preparations do not provide confirmative diagnosis. Tonsil and appendix specimens prepared for other purposes can be analyzed using pathological tests to determine epidemiological statistics.12,13 The results of these tests revealed that the number of asymptomatic but vCJD infected individuals was increasing throughout the general population in the UK; however, these results might not reflect the true epidemiological features.14,15 Blood is the most suitable specimen for this purpose if an adequate test method is to be developed. Many investigators have tried to develop a vCJD diagnostic method using blood samples; however, none of these trials was successful.16–20
The difficulty in the detection of abnormal prion protein isoform (PrPsc) can be attributed mainly to the low concentration of this protein in blood. Accordingly, assuming that the concentration of PrPsc reflects its infectivity, the concentration of PrPsc in blood is assumed to be 1 pg/ml or less. However, in many conditions, there was no stoichiometrical relation between PrPsc concentration and infectivity titre. Further, aggregated PrPsc was assumed to be the infective agent of TSE. According to this assumption, the detection of the aggregate in blood would be the most ideal option for diagnosis, the formation of the aggregate being initiated by a conformational change from PrPc to PrPsc. The protein misfolding cyclic amplification (PMCA) system was used first to test the protein-only hypothesis in vitro.21–23 However, it was reported that some anionic substances were required for the multiplication of PrPsc, and synthetic Poly-A was assumed to be one of the most effective of these anionic compounds.24,25 Few studies have reported results in accordance with the assumption that some RNA molecules can associate with PrPsc to form aggregates.26,27
In contrast to the marked differences between the clinical observations of CJD and other brain diseases, somewhat similar characteristics were reported between these other brain diseases. By this reason, these brain diseases other than CJD have been sometimes suspected to overlap with the prion disease in their disease expression mechanisms. 28 In spite of their clinical differences, these diseases share some biochemical similarities.
We found that weak acidic conditions (inoculation of acidic saline) facilitated the precipitation of PrPsc-containing aggregate from scrapie-infected hamster plasma (acidic SDS precipitation system; ASP). The precipitation reaction induced by the acidic condition was further confirmed using scrapie-infected hamster brain homogenate by the presence of an aggregation partner, which was most probably the RNA containing Poly-A.29,30 Therefore, we evaluated the RNA detection in the plasma samples of spCJD and GSS patients or in the plasma samples of patients suspected to have other brain disorders.
Materials and Methods
Enzymes and chemicals
DNaseI was purchased from Promega Co. Ltd. RNase A (Nakarai chemicals, Co. Ltd) was a gift from Dr. Sato of our laboratory. RNase A inhibitor was purchased from Roche Diagnostics. Proteinase K (PK; 600 mAnson u/mg protein; Merck Co. Ltd), guanidine isothiocyanate (GdnTC; Invitrogen Co. Ltd), isopropyl alcohol (IP; Sigma) and SYBR GII (Invitrogen Co. Ltd) were also purchased. The dye used for denaturing samples, the denaturing gel buffer and the 3-morpholino-propane-sulfonic acid electrophoresis buffer for agarose gel electrophoresis were all purchased from Ambion Co. Ltd.
Plasma samples
All the patients recruited for donating plasma samples were undergoing treatment under YI at Oyamada Memorial Spa Hospital. Informed consent was obtained from these patients and blood samples were collected thereafter. The list of these patients is shown in Table 1. The blood of healthy volunteers recruited from general population was used to prepare the negative control plasma. These healthy volunteers’ blood were donated for the primary purpose of transfusion but had not been used because of inadequate volume. This donated blood samples were used as the negative control. The plasma preparations from the patients and the healthy volunteers were centrifuged at 3,000 rpm for 10 min to separate the test and negative control plasma.
Donor patients of test plasma.

Sporadic CJD.
Gerstmann–Sträussler syndrome.
Multiple system Atrophy.
Parkinson's disease.
Spinocerebellar Degeneration.
Genetic mutation (Val 180 Ile) was determined.
Genetic mutation (Pro 102 Leu) was determined.
not done.
Denaturing solution
Denaturing solution (DN) during the processing of the test plasma was prepared using 5.5 M or 4.0 M GdnTC, 0.5% of N-lauroyl sarcosine sodium salt (Sarcosyl) in 25 mM of a citrate buffer (pH 7.0) and 0.1 M 2-mercaptoethanol. Hereafter, these solutions are referred to 5.5 M DN or 4.0 M DN, respectively.
Processing of plasma samples
The processing method of the plasma samples is presented in Figure 1. In brief, DNase and RNase (20 μ/ml each) were added to 50 μl of a plasma sample and the solution was incubated for 10 min at 37 °C. Subsequently, SDS, dithiothreitol (DTT) and ethylenediaminetetraacetic acid (EDTA) (final concentrations: 3%, 50 mM, and 2 mM, respectively) were added to the solution, mixed and then boiled for 10 min. The mixture was cooled immediately on ice; subsequently, 50 μg/ml of PK was added followed by incubation for 30 min at 37 °C. Furthermore, a tenfold volume of 4.0 or 5.5 M DN and a 1/10 volume of 2 M sodium acetate (NaOAc; pH 4.0) were added to the reaction mixture and mixed vigorously. Next, 300 μl of a mixture of phenol, chloroform and isoamyl alcohol (PCI) was added to the mixture, which was then subjected to vigorous shaking and cooling on ice for 15 min. The chilled mixture was centrifuged for 10 min at 20,000 × g, and then the upper water-soluble layer was collected. To this upper layer, PCI was added again and the mixture was processed by the aforementioned procedure. Then, a 1/20 volume of 3 M NaOAc and a 2.5-fold volume of IP were added to the collected fraction, and the mixture was then cooled for 3 min on ice and centrifuged at the same condition, and the precipitate was collected. The precipitate was then dissolved in 150 μl of 4.0 M DN. Next, a 2.5-fold volume of IP was added without NaOAc; the mixture was centrifuged at the same condition and the precipitate was collected. The precipitate was dissolved in 100 μl of 10 mM Tris pH 7.2 containing 5 mM EDTA and 1% SDS (TES), precipitated using a threefold volume of ethanol (EtOH) and centrifuged for 10 min. This final precipitate was rinsed with a small volume of EtOH, dried, and electrophoresed.

Processing of plasma samples and extraction of RNA. Summary of the RNA extraction procedure from plasma is shown. Although the overall process is similar to the well-known method for RNA extraction from tissue specimens, several minor changes were included as indicated below. (1) RNase and DNase treatment (20 u/ml) of the plasma in the initial step; (2) boiling after the addition of SDS/DTT/SDTA; (3) digestion of the boiled plasma with PK (50 μg/ml) at 37 °C for 30 min; (4) treatment of PK-digested plasma with DN (containing 5.5 M or 4.0 M GdnTC); and (5) approximately 2.5-fold more volumes of IP were used for RNA extraction from the tissue specimen.
Sample analysis
The processed plasma preparations were electrophoresed on 1.5% denatured agarose gel, followed by staining with CYBR GII fluorescence dye; the signals on the stained gel were analyzed using an LAS3000 image analyzer (FUJIFILM Co. Ltd).
Results
Extraction method
It is believed that only a minimum amount of nucleotides exist in the blood, if any. Because of this reason, we had to introduce slight modifications to the protocol of the RNA extraction method which has been described elsewhere (Fig. 1). We have modified the following steps: 1) DNase and RNase treatment were included in the first step to prevent nonspecific adsorption of proteins to free nucleotides; 2) boiling in the presence of SDS and subsequent digestion of most proteins with PK was the second step; and 3) a 2.5-fold greater volume of IP was used in the first and second precipitation steps to obtain the minimum desired amount of RNA.
RNA extraction from plasma samples of patients
We identified two CJD patients (including CJD with codon 180 mutation (genCJD), two non-TSE patients with a neurodegenerative disease and two healthy volunteers on the basis of detecting RNA in the plasma samples. As indicated in Figure 2, SYBR GII staining revealed signals of similar size (1.5–2.0 kb). Furthermore, the signal strength of the samples from the two CJD patients was similar, but the signals from the plasma sample of the non-CJD patients showed some variation. No RNA was detected in the plasma from the healthy volunteers.

Detection of desired RNA from plasma samples of patients with classical CJD and other brain disorders. We used 50 μl of plasma samples from each of the indicated patients or healthy volunteers for the extraction of RNA-like substances. The extraction method was as indicated in Figure 1 and the Materials and Method section. The extraction products were electrophoresed on 1.5% agarose gel. After electrophoresis, the gel was stained using SYBR GII fluorescence dye and the stained signals were detected using the LAS 3000 image analyzer. The plasma samples were obtained from patients (lanes 1–4) or healthy volunteers (lanes 5 and 6); lanes 1: CJD3, 2: CJD4, 3: PD2, 4: SCD, 5: HV1, 6: HV2.
Dependencies of extraction condition
DNA and RNA have distinct pH-dependent solubility in phenol. Therefore, we examined whether it was possible to distinguish between the signals of the plasma samples from diseased and healthy individuals at low pH (Fig. 3A), and if the signal could be obtained at both high and low pH conditions (Fig. 3B). As indicated in Figure 3A, the difference between CJD and healthy individuals was clearly observed in signal recovery. Moreover, signal recovery using 5.5 M DN appeared to be more effective than using 4.0 M DN, although both DNs provided similar results. We further examined if signal recovery at high and low pH conditions allowed us to discriminate between diseased individuals and healthy ones; the low pH (pH 4.0) provided favourable results, whereas thehigh pH (pH 8.4) did not (Fig. 3B). These observations clearly suggest that low pH conditions should be used for obtaining good signal recovery and to facilitate the discrimination of diseased individuals from healthy ones.

Efficiency of extraction of RNA-like substance from the plasma samples of a CJD3 patient. A) Extraction efficiencies of 5.5 M DN and 4.0 M DN were compared using plasma samples of either patient CJD3 or donor HV1. Two tubes of each plasma sample were first treated with nucleases and SDS, and boiled, and then PK was added to the tubes as indicated in Figure 1; subsequently, 5.5 M or 4.0 M DN and 2 M NaOAc (pH 4.0) were added into each tube. These plasma samples were processed according to the routine procedure, and signal recovery for each DN condition was analyzed. Lanes: 1: CJD1, 5.5 M DN; 2: CJD1, 4.0 M DN; 3: HV1, 5.5 M DN; 4: HV1, 4.0 M DN. B) The effect of pH on the DN treating step was evaluated. Plasma samples from patient CJD3 or donor HV1 were processed as mentioned before. To the processed plasma samples, 4.0 M DN and NaOAc of pH 8.4 or 4.0 were added. The subsequent procedure was the same as described in Panel A. Lanes: 1: CJD1, pH 8.4;2: CJD1, pH 4.0;3: HV1, pH 8.4;4: HV1, pH 4.0.
RNase sensitivity of the extracted signal
Because the plasma samples of CJD patients obtained after the PCI step distinctly revealed a signal in low pH condition, this signal was strongly suspected to be RNA. Therefore, we determined the sensitivity of the signal to nucleases (Figure 4). After PCI treatment, the precipitates obtained from the plasma samples of CJD patients and healthy individuals were washed once using TES containing no additives (none), RNase inhibitor (RNI), RNase A (RNase) or DNase 1 (DNase), and finally precipitated using EtOH. The EtOH precipitates were dissolved using the same solvent as used in the former washing step. The dissolved preparations were then incubated at 37 °C for 10 min but the preparations dissolved in TES with no additive were retained on ice. Because even the incubation at 37 °C degraded the RNA in the preparation to some extent, the retained preparations without further additives and no incubation showed the original amounts. As shown in Figure 4, RNase treatment affected the signal, although to a small extent, whereas RNI or DNase treatment showed no effect on the signal.
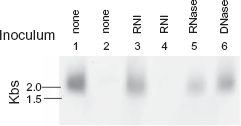
Effect of nucleases on the extracted substance. The extract from patient CJD3 or the HV1 control plasma sample was washed once with TES containing the test substances, and subsequently dissolved in TES containing RNase inhibitor, RNase or DNase. These preparations were incubated at 37 °C for 10 min and electrophoresed on 1.5% agarose. Substances from the same amount of starting plasmas were retained untreated on ice before electrophoresis. A part of the extract from the CJD 3 or HV1 plasmas were retained untreated on ice to offer the negative control for incubation effect at 37 °C. Lanes: plasma extract from 1: CJD3, untreated, 2: HV1, untreated, 3: CJD3, RNase inhibitor (RNI) treated; 4: HV1, RNI treated; 5: CJD3, RNase treated; 6: CJD3, DNase treated.
Signal detection in patients with different brain disorders
To determine if RNA signal could be repeatedly obtained from patients with brain diseases, plasma samples from patients with CJD, GSS and other brain diseases were compared with the plasma samples from two healthy volunteers by examining at the extraction condition using 5.5 M DN (Fig. 5: Panel A). Plasma samples from the 10 healthy individuals and from two spCJD patients were also examined under the same condition in reverse (Fig. 5: Panel B). As indicated in Fig. 5, RNA-like signals were observed in all the examined patients’ plasma samples, although low intensity signals were obtained from plasma samples of patients PD2, SCD and PD3 in 5.5 M DN solution (Fig. 5: Panel A; see Table 1 for patient labels). In comparison with this observation, negligible signals were obtained from the plasma samples of healthy individuals (Fig 5: Panel A; lanes 11 and 12, Panel B; lanes 3–12). As shown in this figure, the plasma samples from CJD2 and CJD3 patients yielded signals of good intensity. This result confirmed that the signal was not an artefact.
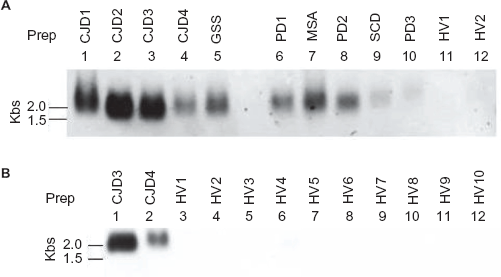
Detection of signals in the plasma extract of patients with CJD, GSS, other brain disorders or healthy individuals. A) The plasma samples of patients with CJD, GSS and other brain disorders were processed for RNA extraction. The method of processing is described in the Materials and Methods section. The upper panel was extracted using 4.0 M GdnTC at the denaturing step (4.0 M DN). The lower panel was extracted using 5.5 M GdnTC at the denaturing step (5.5 M DN). The lanes are indicated in each panel. The plasma samples from two healthy volunteers (HV1 and HV2) were examined simultaneously for the negative control on the RNA extraction in plasma. B) The RNA extract from plasma samples of healthy volunteers was examined and compared with the extract obtained from the plasma of patients with CJD, GSS or other brain disorders. RNA was extracted from the plasma samples of 10 healthy volunteers and two CJD patients (CJD3 and CJD4) using 5.5 M DN. The relevant lanes are indicated in the panel.
Discussion
We attempted to develop a blood test for the diagnosis of classical CJD based on the detection of PrPsc from plasma using a formally developed ASP method to discriminate CJD patients from healthy individuals. This study was planned on the basis of the consensus that PrPsc was a confirmative marker of TSE. However, we were unable to obtain satisfying results.
Recent studies on the properties of the infectious agent revealed that a 25 nm virus-like particle was the most likely infectious agent.30,31 These findings indicated that the infectious agent of scrapie was not a large fibril but a relatively small-sized particle containing a much smaller amount of PrPsc. There was no definitive conclusion regarding which argument was more correct for the present problem. Therefore, we were unable to establish a definite target of detection for the blood test. For this purpose, investigation of both PrPsc aggregates and other novel non-PrPsc markers in blood was warranted.
Because many difficulties were encountered during the development of the blood test, subsequent efforts were directed toward detecting the presence of aggregates containing PrPsc in blood. Subsequently, many methods were proposed. These methods have tendencies to prpose the antibody-capture immunoassay that employs PrPsc-aggregate specific monoclonal antibodies. The aggregates were detected by 1) fluorescence detection of DNA coupled with anti-PrPsc antibody, 2) by laser scanning or 3) by enhanced polymerization by inoculating the recombinant seed and detection of the resulting amyloid.19,32,33 In addition, the PMCA method was also applied to develop a blood test as this was expected to be the most applicable one. 34 Detection of small amount of PrPsc in blood with high specificity could be achieved by in vitro multiplication of PrPsc. However, this method could be used only with normal brain homogenate as the PrPsc substrate (platelets or other cell homogenates were later proved to be suitable for use). Furthermore, this disadvantage was more serious in the presence of poly-anions (especially synthetic poly-A RNA), which are necessary for effective in vitro multiplication of PrPsc.24,25 Moreover, plasma proteins inhibited in vitro multiplication of PrPsc. On the basis of these disadvantages, the use of this method in the blood test was disputed. Therefore, developing a method for a blood test for diagnosing CJD (spCJD and vCJD) by detecting PrPsc in blood samples was encumbered with difficulties.
To solve this problem, it was first essential to confirm the nature of the specific marker of TSE and then develop a blood test. In addition to the observation that poly-anions were necessary for the in vitro multiplication of PrPsc, several pieces of evidences suggested that PrPsc exhibited high affinity toward nucleic acid.26,27 Hence, TSE-infected individuals could have nucleic acid in their blood. Recently, two small-sized RNA molecules were identified in a scrapie- associated fibril (SAF) from scrapie-infected sheep brain; whether this report indicated the real infective agent to the RNA–PrPsc complex remains to be clarified. But the possibility that the infective agent contained nucleic acid was increased. (Simoneau et al 2009, unpublished data.) It is possible that nucleic acid could be a component of the PrPsc-containing aggregate in blood. Therefore, it is necessary clarify if the infectious 25 nm particles contain some nucleic acid in addition to the PrPsc protein.
We reported previously that poly-A RNA might be an important partner for the aggregate containing PrPsc as determined by the ASP method. 29 This observation supported the finding of the two aforementioned reports24,25 that poly-A RNA might play an important role in in vitro multiplication of PrPsc in the PMCA method and that RNA may be the necessary requirement for SAF infectivity.24,25 Therefore, it was important to confirm the presence of any RNA in the blood of the patient. As an alternative to detecting PrPsc in the blood sample, which involved a very difficult procedure, if we could detect RNA in the blood, this could be a desirable for the development of a blood test. In this context, we examined the presence of RNA in the blood samples of spCJD and GSS patients. Figure 2 shows the existence of a SYBR GreenII-stained component, which is approximately 1.5–2.0 kb in size, in the plasma sample of a patient. This substance may be RNA because it was extracted under acidic conditions and was RNase sensitive, although it was not digested completely after RNase treatment (Figs. 3 and 4). Because this RNA was not detected in the plasma sample of a healthy volunteer, it can be considered to be firmly associated with the diseased state. Therefore, this RNA could be considered as a potential marker of spCJD and GSS. As mere incubation degraded the RNA to some extent as indicated (Fig. 4 lanes 1 and 3), longer incubation could not be carried out. In addition, minimal amounts of remainng GdnTC from the DN together with the SDS in the reaction buffer (TES) may cause the prevention of complete digestion of RNA in this reaction.
We also examined the presence of RNA in the plasma of patients with non-TSE brain disorders. These brain disorders were also suspected to be correlated with the prion diseases within the disease expression although their non-transmissible nature was clinically proved.35–39 In these reports, suspected transmissibility of Parkinson's disease (PD), amyloid precursor protein(APP) processing and β-amyloid deposition in a spCJD patient were observed. From these observations, the possible existence of overlapped common processes through disease expression between the amyloidosis was assumed.36,38–42 We found RNA fractions of similar size in patients MSA, PD1, PD2, PD3 and SCD, which have the possibility to become the marker of the common process. Among the patients, those with GSS and PD exhibited variation in the amount of detectable RNA, unlike CJD patients.
Only a small number of patients could be tested here, but the existence of the RNA in the plasma sample of the patients’ plasma should be stressed. We could not test a greater number of patients as we do not have any more patients being treated for these conditions at our hospital. It is important to confirm the observation indicated here by testing the number of patients for the presence of RNA in the plasma.
Analysis of the properties of this RNA was most important. As mentioned before, recent reports suggest that small-sized RNA fragments (27 and 54 bases) were important constituents of SAF observed in scrapie-infected sheep brain. (Simoneau et al 2009, unpublished data.) However, the extraction procedure and the size of RNA obtained in this study showed marked differences from ours; therefore, their RNA in the SAF might be different from that identified in our study. In addition, we cannot compare RNA in SAF samples obtained from the brain and those from the plasma, even if we extract both by our method. In spite of these differences, the observations that SAF originating from the scrapie-infected hamster brain contained RNAs and the PrPsc-RNA complex acquired infectivity suggested the infective agent of the TSE was believed to be composed of the PrPsc protein only in the protein only hypothesis. If SAF which was the concentrate of the infectivity contained some nucleic acid as the important constituent of infective agent it became the direct evidence to indicate the fault of the protein hypothesis.
This may be one of the greatest arguments of the protein-only hypothesis. Although the RNA isolated from the plasma samples of patients in our study was not confirmed to show infectivity, it could become an important marker for discrimination between patients with spCJD, GSS or other brain diseases, and healthy individuals. Comparative analysis of these two RNAs, namely the RNA found in the SAF and that identified in our study, should be carried out. In addition, it is also important to determine whether these RNAs were the real components of the infectious agent of TSE.
Furthermore, it is also important that a overlapping common marker has possibly been identified for detecting brain disorders such as spCJD, GSS and non-TSE brain diseases. In this regard, we have been successful in confirming the existence of a common marker in the plasma of patients with spCJD and other disorders. Clinically, the existence of common RNA suggested a possibility that some of these brain diseases should be categorized as a group of disease which share the expression of some common markers. In order to solve these problems, the nature of this RNA must be clarified immediately.
We evaluated the diagnostic importance of detection of nucleotides in plasma. Appearance of nucleotides in plasma is an important marker of several diseases. However, it has been difficult to detect the presence of nucleotides until now. The reason for this failure was unknown and its further investigation was discouraged. 43 In addition to its application in diagnosing TSE, the RNA extraction method described here can also be used for other purposes to obtain nucleotides in plasma.
Abbreviations
Transmissible spongiform encephalopathy;
Disease associated isoform of prion protein;
Protein misfolding cyclic amplification;
Proteinase K;
Guanidine isothiocyanate;
Denaturing solution;
Sodium acetate;
Mixture of phenol/chloroform/isoamyl alcohol;
Isopropyl alcohol;
Ethanol;
Acidic SDS precipitation;
10 mM Tris pH 7.2 containing 5 mM EDTA and 1% SDS;
CJD with codon 180 mutation;
dithiothreitol SDS: Sodium Lauryl Sulfate;
sporadic Creutzfeldt–Jakob disease;
variant Creutzfeldt–Jakob disease;
Gerstmann–Straüssler syndrome;
ethylenediaminetetra-acetic;
abnormal prion protein isoform;
Parkinson's disease.
Publish with Libertas Academica and every scientist working in your field can read your article
“I would like to say that this is the most author-friendly editing process I have experienced in over 150 publications. Thank you most sincerely.”
“The communication between your staff and me has been terrific. Whenever progress is made with the manuscript, I receive notice. Quite honestly, I've never had such complete communication with a journal.”
“LA is different, and hopefully represents a kind of scientific publication machinery that removes the hurdles from free flow of scientific thought.”
Available to your entire community free of charge
Fairly and quickly peer reviewed
Yours! You retain copyright
Footnotes
Acknowledgment
First, we are very grateful to our patients in Oyamada Memorial Spa Hospital who offered their blood. Even though these patients were unwell and in bed, they kindly agreed to participate in the study by providing their blood for investigation. We thank Dr. Sato from our institute for his discussions and the kind gift of RNase. We also want to thank Drs. Satake, Uchida and Nishimura for their discussions. We thank all the members of Oyamada Memorial Spa Hospital and all members of the Japanese Red Cross society (JRC) for their support.
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.