
Abstract
Expression and secretion of apolipoprotein A-I (apoA-I) by cultured liver cells can be markedly stimulated by triazolodiazepines (TZDs). It has been shown previously that the thieno-TZD Ro 11-1464 increases plasma levels of apoA-I and in vivomacrophage reverse cholesterol transport in mice. However, these effects were only seen at high doses, at which the compound could act on central benzodiazepine (BZD) receptors or platelet activating factor (PAF) receptors, interfering with its potential utility. In this work, we describe 2 new thieno-TZDs MDCO-3770 and MDCO-3783, both derived from Ro 11-1464. These compounds display the same high efficacy on apoA-I production, metabolic stability, and lack of cytotoxicity in cultured hepatocytes as Ro 11-1464, but they do not bind to the central BZD receptor and PAF receptor. The quinazoline RVX-208 was less efficacious in stimulating apoA-I production and displayed signs of cytotoxicity. Certain TZDs stimulating apoA-I production are now known to be inhibitors of bromodomain (BRD) extra-terminal (BET) proteins BRDT, BRD2, BRD3, and BRD4, and this inhibition was inferred as a main molecular mechanism for their effect on apoA-I expression. We show here that the thieno-TZD (+)-JQ1, a potent BET inhibitor, strongly stimulated apoA-I production in Hep-G2 cells, but that its enantiomer (-)-JQ1, which has no BET inhibitor activity, also showed considerable effect on apoA-I production. MDCO-3770 and MDCO-3783 also inhibited BRD3 and BRD4 in vitro, with potency somewhat below that of (+)-JQ1. We conclude that the effect of thieno-TZDs on apoA-I expression is not due to inhibition of the BZD or PAF receptors and is not completely explained by transcriptional repression by BET proteins.
Introduction
Several independent lines of research have shown that direct administration of High Density Lipoprotein (HDL) or hepatic transgenic overexpression of its main protein apolipoprotein A-I (apoA-I) can rapidly produce plaque regression in animal models of atherosclerosis.1–5 Thus, upregulation of apoA-I production might be beneficial for treatment of this disease. Previously, using cell-based assay systems, triazolodiazepines (TZDs) were discovered, which are able to enhance apoA-I gene expression and protein synthesis/secretion in cultured liver cells or Hep-G2 cells. These comprise members of thieno-TZDs such as Ro 11-1464, 6 the benzo-TZDs U-34599, and U-51477, 7 as well as GW841819X, 8 all reported to stimulate apoA-I production 4- to 6-fold at micromolar concentrations (structures of these compound shown in Fig. 1). Recently, we showed that Ro 11-1464 increased plasma levels of apoA-I in mice transgenic for human apoA-I. 9
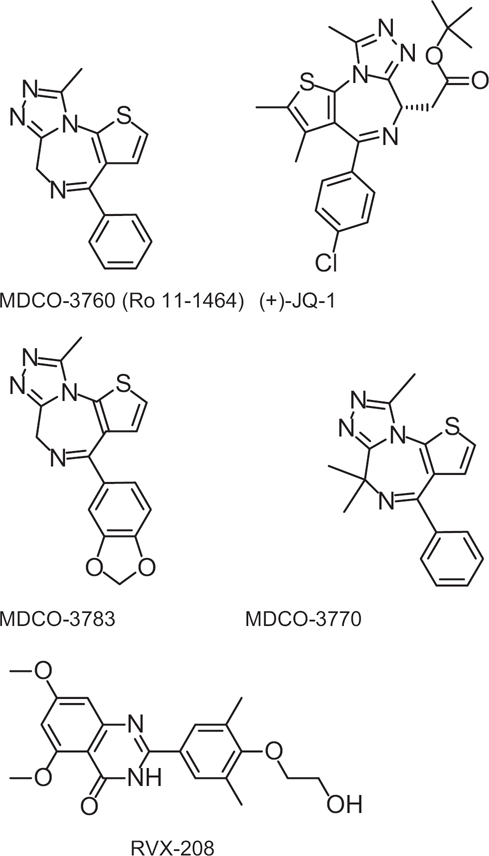
Structures of compounds tested in this study.
TZDs were developed in the past as sedatives acting as agonist on the benzodiazepine (BZD) receptor in the central nervous system (eg, triazolam, brotizolam) or as antiplatelet agents acting as antagonists of the platelet-activating factor (PAF) receptor.10,11 However, whether the stimulating effect of TZDs on apoA-I expression is mediated by binding of these TZDs to the central BZD receptor and/or to the PAF receptor has not been demonstrated. The first aim of this paper, therefore, is to describe synthesis and characterisation of 2 novel thieno-TZDs derived from Ro 11-1464, which are completely free of binding to these receptors but nevertheless remain good apoA-I upregulators.
Researchers at Resverlogix Inc have described novel quinazolines as upregulators of apoA-I. Their most advanced compound, RVX-208, stimulates apoA-I protein production by HepG2 cells 2-fold at 40 μM. 12 The second aim of our work was to compare our thieno-TZDs with RVX-208 with respect to apoA-I production and potential cytotoxicity.
Recent work by GlaxoSmithKline investigators ascribed the activity of certain benzo-TZDs on apoA-I expression to displacement of bromodomain and extraterminal (BET) proteins from acetylated histones. 8 Likewise, other TZDs were recently established as potent and selective BET inhibitors, that is, the thieno-TZD (+)-JQ1, 13 the benzo-TZD GSK525762A (also called I-BET), 14 and the benzotriazolotriazepine BzT-7. 15 The third purpose of this work, therefore, was to establish a comparison of our thieno-TZDs with these recently described thieno-TZDs with respect to apoA-I upregulation and establish if the effect on apoA-I production correlated with BET binding and inhibition.
Our findings show that our new thieno-TZDs are effective upregulators of apoA-I production in liver cells without cytotoxic effects and act independently of binding to central BZD or PAF receptors.
These thieno-TZDs are able to bind and inhibit BET, but additional mechanisms may be involved in their effect on apoA-I expression.
Materials and Methods
Compounds
The structures of the compounds studied in this paper are depicted in Figure 1. Synthesis of the thieno-TZDs MDCO-3760 (Ro 11-1464), MDCO-3783, and MDCO-3770 were described in detail previously. 16 RVX-208 was synthesized as described by Hansen et al. 17 In addition, (+)-JQ1 and (-)-JQ1 were synthesized as described. 12
Testing of compounds in cultured hepatocytes and Hep-G2 cells
Source of hepatocytes, culture conditions
Primary human hepatocytes (Ready Heps™ Fresh Human Hepatocytes) as well as the appropriate cell culture medium (HCM™ BulletKit™) were obtained from Lonza Cologne GmbH, Cologne, Germany. The cells were delivered in 96-well platescoated with human collagen at a density of ~5 × 105/well. Upon arrival, cells were handled according to the manufacturer's instructions: briefly, plates were immediately unpacked, the transport film was removed, and cells were allowed to recover for 2 hours at 37°C in 5% CO2 in air. Following recovery, transport medium was removed and replaced by 200 μL/well prewarmed HCM™ medium. The HCM™ medium was prepared by addtion of SingleQuots™ supplements and growth factors to HBM™ medium, the 2 components of the HCM™ BulletKit™. Cells were processed immediately after recovery.
On the day of the experiment, supernatant medium was discarded and replaced by a solution of test compounds in HCM™ medium, which was prepared from DMSO stock solutions immediately before use. Stock solution concentrations assured that DMSO never exceeded 0.1% in the assay, as DMSO alone exerts significant effects on hepatocyte gene expression. Each concentration was run in triplicate whereas multiple wells were prepared for vehicle (DMSO) controls. The cells were cultured in the presence of the studied compounds and DMSO control for 72 hours at 37°C in 5% CO2 in air. The supernatants were replaced by a fresh dilution of test compounds after 24 hours and 48 hours, respectively. Supernatants were collected and stored at −80°C until further analysis.
The human hepatocellular carcinoma cell line HepG2 was obtained from the DMSZ. HepG2 cells were routinely cultured in 75 cm2 flasks in DMEM medium supplemented with 10% FCS, 1% penicillin/streptomycin at 37°C in 5% CO2 in air. Cells were split every 3 to 4 days at 80% confluence. HepG2 cells were seeded into 96-well plates at a density of 2.5 × 105 cells/well and were incubated overnight prior to starting the assay. Experiments were then conducted as described above for primary human hepatocytes.
ApoA-I quantitation by enzyme-linked immunosorbent assay (ELISA)
After removal of the supernatants after 72 hours, all wells were washed 3 times with 200 μL isotonic 0.29 MD-mannitol and 25 μL d H2O was added to each well. The protein content of each well was determined using the Pierce® BCA Protein Assay Kit according to kit instructions and calculated as micrograms cellular protein per well.
For apoA-I detection, the human apoA1 ELISA kit from Mabtech Nacka, Sweden (Catalog no. 3710-1H-20) was used. Microplates were coated with 100 μL/well of the monoclonal antibody HDL 110 (diluted to 2 μg/mL in PBS), incubated at room temperature overnight, then washed twice with 200 μL/well PBS, and blocked for 1 hour by addition of 200 μL/well incubation buffer containing PBS, 0.05% Tween 20 and 0.1% BSA. Plates were then washed 5 times with washing buffer containing PBS/0.05% Tween 20, and 100 μL/well of diluted cell culture supernatant or standards were added. A standard curve was prepared in the concentration range between 0.1 and 100 ng/mL. Next, 100 μL/well of analyte or standard was added to duplicate wells, and the plates were incubated for 2 hours at room temperature. Subsequently, the plates were washed 5 times with 200 μL/well washing buffer, 100 μL/well of the biotinylated monoclonal antibody HDL44, at 0.5 μg/mL in incubation buffer, were added, and the plate was incubated for 1 hour at room temperature. Following another washing step, 100 μL/well of Streptavidin-HRP diluted 1:1000 in incubation buffer was added, and the plates were incubated for 1 hour at room temperature. Finally, the chromogenic substrate was added, and absorbance was measured at 450 nm.
The apoA-I content of each sample was calculated using the standard curve generated with each plate and was expressed as ng apoA-I per mg of cellular protein.
Cytotoxicity parameters (mitochondrial potential and ATP level)
HepG2 cells (at a density of 25 × 103 cells per 100 μL) and HT1080 cells (human fibrosarcoma cells, at a density of 25 × 103 cells per 100 μL) were cultivated in DMEM with 10% FCS. After incubating the cells in 96-well microplates at 37°C and 5% CO2 overnight, test compounds or vehicle (DMEM medium) were added, and the cells were further incubated for 72 hours. Cells were then washed 2 times with HBSS before cytotoxicity was analyzed by one of the following methods. (1) The ATP content of the cells was measured using the Cell Titer Glo assay from Promega Mannheim, Germany according to kit instructions. Briefly, 100 μL of cell culture medium and 100 μL of the Cell Titer Glo Reagent (freshly reconstituted Cell Titer Glo Substrat and Cell Titer Glo buffer) were added to each well. After mixing and incubating the plate at room temperature for 10 minutes, luminescence was recorded using a FLUOstar optima plate reader from BMG Labtech Jena, Germany. The luminescence signal generated is proportional to the amount of ATP released from lysed cells. (2) For measurement of the mitochondrial membrane potential, 100 μL of a TMRM (tetramethylrhodamine methyl ester) solution (1 μM in culture medium) was added to the wells, and the plates were incubated for another 30 minutes at 37°C and 5% CO2. Following 3 washing steps with HBSS, fluorescence intensity was measured at 590 nm (excitation at 544 nm).
Metabolic stability and uptake of compounds by liver cells
Cryopreserved human hepatocytes from BD Biosciences, Heidelberg, Germany (catalog number 454503) and from 2 different donors were used. Prior to use, hepatocytes were purified using the Hepatocyte purification kit (BD Biosciences, Heidelberg, Germany catalog number 454600) according to the manufacturer's protocol. The cell concentration was adjusted to 1 − 106 cells/ml in Krebs-Henseleit (K-H) buffer (Sigma-Aldrich Taufkirchen, Germany). To 125 μL of cell suspension, 120 μL K-H buffer and 5 μL of a test compound solution in distilled water (final concentration 10 μM) were added. Reaction mixtures were incubated with gentle shaking at 37°C in a water bath. For protein precipitation and cell lysis, the samples were mixed with 250 μL ice-cold acetonitrile after 0 (control), 60, and 120 minutes, respectively. After 10 minutes at room temperature, the samples were centrifuged at 14,000 rpm for 15 minutes. Next, 220 μL of the supernatant was evaporated to dryness in a stream of nitrogen. The residue was dissolved in 100 μL distilled water and analyzed by HPLC. All experiments were performed in duplicate.
Binding to BZD and PAF receptors
Binding affinities of MDCO-3760, MDCO-3770, and MDCO-3783 to the central BZD receptor were measured in rat cerebral cortex membranes, using 3H-flunitrazepam as ligand. Diazepam used, as the reference has an IC50 of 20 nM. For PAF binding, the human recombinant PAF receptor was expressed in CHO cells, and binding was measured as inhibition of 3H-C16-PAF to these cells. Unlabeled C16-PAF used as reference has an IC50 of 0.7 nM.
Bromodomain binding measurements
Protein stability shift assay
Thermal melting experiments were carried out using an M x 3005p Real Time PCR machine (Stratagene Waldbronn, Germany). Proteins were buffered in 10 mM HEPES pH 7.5, 500 mM NaCl and assayed in a 96-well plate at a final concentration of 2 μM in 20 μL volume. Compounds were added at a final concentration of 10 or 100 μM. SYPRO Orange (Molecular Probes Darmstadt, Germany) was added as a fluorescence probe at a dilution of 1 in 1000. Excitation and emission filters for the SYPRO-Orange dye were set to 465 nm and 590 nm, respectively. The temperature was raised with a step of 3°C per minute from 25°C to 96°C, and fluorescence readings were taken at each interval. The temperature dependence of the fluorescence during the protein denaturation process was approximated by the equation
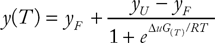
Isothermal titration calorimetry
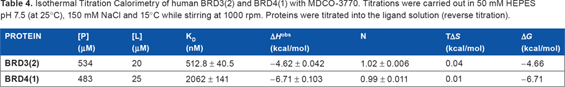
Experiments were carried out on an ITC200 titration microcalorimeter from MicroCal™, (GE Healthcare) Little Chalfont, Bucks, UK equipped with a washing module, with a cell volume of 0.2003 mL and a 40 μL microsyringe. Experiments were carried out at 15°C while stirring at 1000 rpm, in ITC buffer (50 mM HEPES pH 7.5 (at 25°C), 150 mM NaCl). The microsyringe was loaded with a solution of the protein sample (534 or 483 μM protein for BRD3[2] and BRD4[1] respectively, in ITC buffer) and was carefully inserted into the calorimetric cell, which was filled with an amount of the ligand (0.2 mL, 20–25 μM in ITC buffer). The system was first allowed to equilibrate until the cell temperature reached 15°C, and an additional delay of 60 seconds was applied. All titrations were conducted using an initial control injection of 0.3 μL followed by 38 identical injections of 1 μL with a duration of 2 seconds (per injection) and a spacing of 120 seconds between injections. The titration experiments were designed to ensure complete saturation of the proteins before the final injection. The heat of dilution for the proteins was independent of their concentration and corresponded to the heat observed from the last injection following saturation of ligand binding, thus facilitating the estimation of the baseline of each titration from the last injection. The collected data were corrected for protein heats of dilution (measured on separate experiments by titrating the proteins into ITC buffer) and deconvoluted using the MicroCal™ Origin software supplied with the instrument to yield enthalpies of binding (ΔH) and binding constants (KB) using the method previously described in detail by Wiseman and coworkers. 19 Thermodynamic parameters were calculated using the basic equation of thermodynamics (ΔG = ΔH - TΔS = -RTln KB, where ΔG, ΔH, and ΔS are the changes in free energy, enthalpy, and entropy of binding respectively). In all cases, a single binding site model was employed, as supplied with the MicroCal™ Origin software package. Dissociation constants and thermodynamic parameters are listed in Table 4.
Bromodomain inhibition assay (Alphascreen)
Alpha-Screen assays were performed as described previously. 20 A 12-point 1:2 serial dilution of the ligands was prepared to provide a 0–250 μM final assay concentration range. YSGRGK(Ac)GGK(Ac)GLGK(Ac)GGAK(Ac)RHRK (Biotin) peptide was used in the assays.
Buffer conditions used in all experiments were 25 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% BSA, supplemented with 0.05% CHAPS.
Results
Efficacy, selectivity, stability of thieno-TZDs as compared with RVX-208
The first goal of this study was assess the potency and efficacy of 2 new thieno-TZD in comparison with the previously characterized TZD Ro 11-1464 as well as to the quinazoline compound RVX-208 in stimulating apoA-I production by human liver cells. As shown in Figure 2, the original lead compound Ro 11-1464 (denoted here as MDCO-3760) as well as the novel thieno-TZDs derived from Ro 11-1464 (MDCO-3783, MDCO-3770) stimulated apoA-I production by cultured human hepatocytes by up to 3-fold, with similar potency range. In contrast, RVX-208 only achieved up to 80% increase at the highest concentration tested (100 μM).
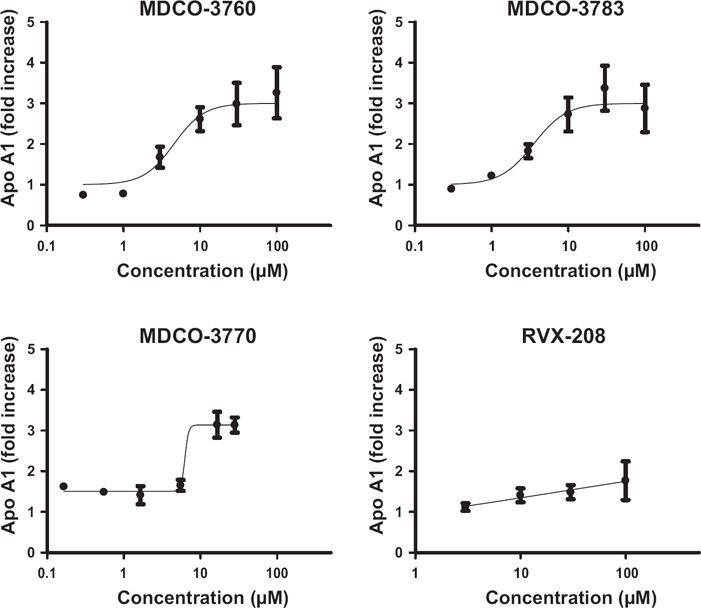
Effect of “MDCO”-thieno-TZDs and of RVX-208 on apoA-I production by primary human hepatocytes (means ± SD for 4 different human hepatocyte cultures).
Since thieno-TZDs are known to act on the central BZD receptor and on the PAF receptor, we assessed the capacity of the above compounds to displace established ligands from these receptors.
As shown in Figure 3, Ro 11-1464 was able to compete with labeled flunitrazepam for binding to rat brain membranes and with labeled PAF for binding to cells expressing the human PAF receptor. In contrast, MDCO-3783 and MDCO-3770 did not compete with labeled flunitrazepam and only weakly displaced labeled PAF from the PAF receptor.
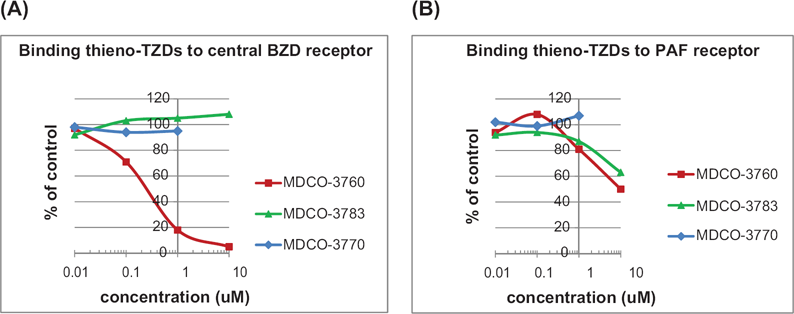
Binding of “MDCO”-thieno-TZDs to central BZD receptor (A) and PAF receptor (B). Binding assays were performed as described in Materials and Methods. Data points are means of duplicates not differing more than 10% of each other.
Since the study compounds are intended to exert a therapeutic effect in liver cells, a further goal of this study was to obtain information with regard to their metabolic stability in liver cells, as well as their effect on indicators of cell viability. As shown in Table 1, the above-mentioned thieno-TZDs are metabolically stable when incubated for 2 hours with human liver cells. As shown in Table 2, these thieno-TZDs did not affect ATP levels in human hepatocytes or the mitochondrial membrane potential in fibrosarcoma cells in concentrations up to 100 μM. In contrast, RVX-208 decreased ATP levels in human hepatocytes by 50% at 100 μM and decreased mitochondrial membrane potential in fibrosarcoma cells with an IC50 of 50 μM.
Metabolic stability of thieno-TZDs in cultured human hepatocytes.

Effect of MDCO-TZDs and of RVX-208 on parameters of cell viability.
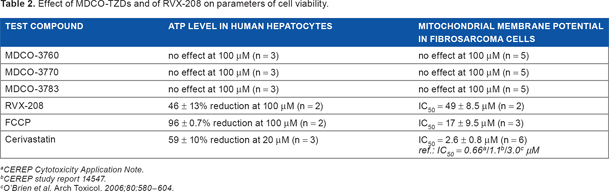
CEREP Cytotoxicity Application Note.
CEREP study report 14547.
O'Brien et al. Arch Toxicol. 2006;80:580–604.
Characterization of thieno-TZDs as BRD binders/ inhibitors
As outlined in the Introduction, related thieno- and benzo-TZDs were recently described to stimulate apoA-I production by cultured liver cells via epigenetic modulation of gene expression, specifically acting as inhibitors of bromodomain proteins.12–14 We therefore set out to establish the ability of our thieno-TZDs as bromodomain ligands and inhibitors, in comparison with the established bromodomain inhibitor (+)-JQ1.
As shown in Table 3, in the protein stability shift assay MDCO-compounds and RVX-208 displayed weaker binding than (+)-JQ1 to bromodomain of BRD2, BRD3, BRD4, and BRDT. RVX208 showed a somewhat stronger binding to the second as compared with the first bromodomain of these BRDs. MDCO-3770 seemed more potent than the 2 other MDCO compounds. For this reason, this compound was selected for isothermal titration calorimetry. The results of that measurement showed strong (2 μM for the first bromodomain in BRD4 and 0.5 μM for the second bromodomain in BRD3) binding affinity (Table 4).
Thermal shift (ΔTmobs) of human bromodomains in the presence of 10 μM compound.
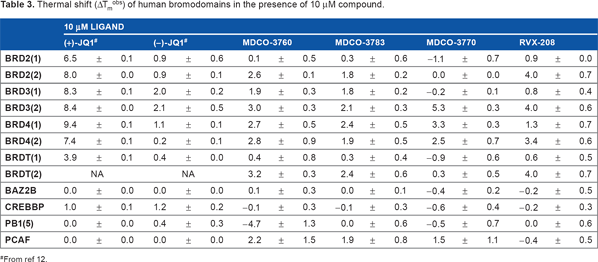
From ref 12.
Isothermal Titration Calorimetry of human BRD3(2) and BRD4(1) with MDCO-3770. Titrations were carried out in 50 mM HEPES pH 7.5 (at 25°C), 150 mM NaCl and 15°C while stirring at 1000 rpm. Proteins were titrated into the ligand solution (reverse titration).
We next assessed the ability of our thieno-TZDs to inhibit bromodomain binding to histone protein domains by using the Alphascreen. 19 As shown in Figure 4, MDCO compounds were clearly able to inhibit binding of an acetylated histone H4 peptide to recombinant BRD3 and BRD4 domains, with a marked preference for the second as compared with the first domain in these BRD proteins. MDCO-3770 was clearly more potent than the other 2 thieno-TZDs, in line with its higher binding affinity observed in the protein stability shift and isothermal titration calorimetry measurements.
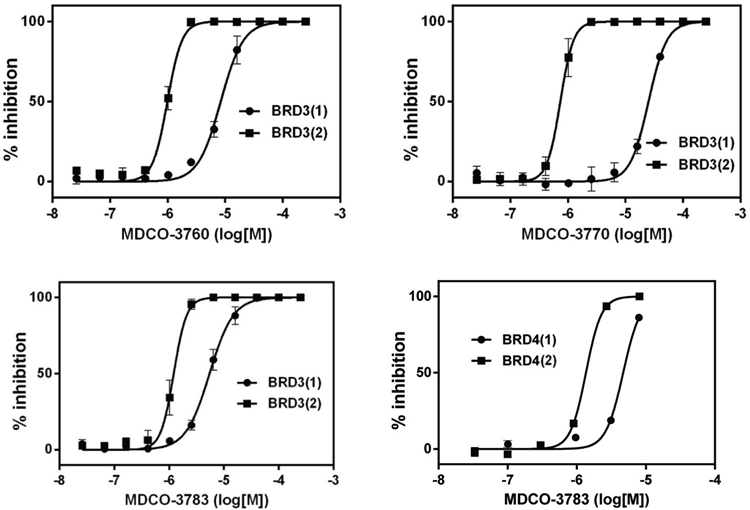
Competitive displacement of binding of acetylated histone 4 peptides to Bromodomain 3 or Bromodomain 4 domains 1 or 2 by MDCO-thieno-TZDs.
Having established that our thieno-TZD are valid bromodomain inhibitors, albeit weaker than the reference compound (+)-JQ1, we then were interested to compare the potency and efficacy of (+)-JQ1 and its inactive enantiomer (-)-JQ1 on apoA-I production with that of one of our thieno-TZDs in Hep-G2 cells. As shown in Figure 5, the potent BET inhibitor (+)-JQ1 was a very effective stimulator of apoA-I production, while significant but lower stimulation was obtained with (-)-JQ1, which is known to be a much weaker BET inhibitor. 13 MDCO-3783 also stimulated apoA-I production by Hep-G2 cells, with a potency and efficacy comparable to that of (-)-JQ1.
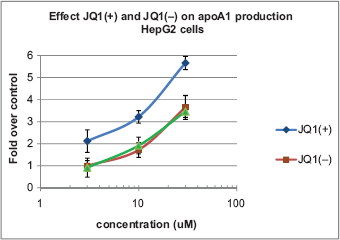
Effect of thieno-TZDs (+)-JQ1, (-)-JQ1, or MDCO-3783 on apoA-I production by HepG2 cells (means ± SD, n = 3).
Discussion
In this paper, we show that the new thieno-TZDs derivatives MDCO-3770 and MDCO-3783 stimulate apoA-I production by human hepatocytes with a potency and efficacy similar to the old scaffold Ro 11-1464 and more strongly than the quinazoline RVX-208.
MDCO-3770 and MDCO-3783 were unable to compete with labeled flunitrazepam for binding to rat brain membranes suggesting that they do not bind to the central BZD receptor, whereas the original lead MDCO-3760 had a definite displacement capacity to BZD and PAF receptor. Together these results show that our thieno-TZDs do not seem to act via these receptors, which were previously established as molecular targets for TZDs.
The MDCO-TZDs were stable during a 2-hour incubation with human hepatocytes and did not affect metabolism (as indicated by ATP) nor mitochondrial membrane potential at concentrations up to 100 μM, suggesting that these com pounds do not affect cell viability. In comparison, RVX-208 affected these cell viability parameters at higher concentrations, in line with signs of liver damage reported in some of the participants in a human study with this compound. 21
Since MDCO-3770 and MDCO-3783 were attractive in terms of effects on apoA-I production and selectivity against other receptors known to bind benzodiazepines and since certain TZD apoA-I inducers were recently described to be BET inhibitors, 8 we decided to characterize our compounds in more detail with respect to their BET binding and inhibitor properties. We also compared the effect of MDCO-3783 with that of the potent BET-inhibitor (+)-JQ1 and its enantiomer (-)-JQ1 on apoA-I production by HepG2 cells. The strong stimulation by (+)-JQ1 could be expected based on the effects of similar TZDs on apoA-I production reported previously. 8
The above-mentioned thieno-TZDs are only weakly binding to BRDs (thermal shift measurements, isothermal titration calorimetry), much weaker than the reference compound (+)-JQ1.
In the BRD inhibition assay, consistent preferences for inhibition of binding to the second domains in BRD3 and BRD4 were seen for all MDCO-thieno-TZDs. Potency for inhibition was highest for MDCO-3770, in line with its higher activity in the protein stability shift assay.
Overall, however, the efficacy or potency of the MDCO and JQ1 TZD compounds to stimulate apoA-I production (Figs. 2 and 5) was not clearly correlated to their binding/competitive inhibition to any of the studied BRDs (Table 3 and Fig. 4). This suggests that in addition to BRD inhibition other yet unknown mechanisms unrelated to BET inhibition are involved in the upregulation of apoA-I production by thieno-TZDs. The observation that the enantiomer (-)-JQ1 also had a noticeable effect, in contrast to the (-) enantiomers of the benzo-TZDs tested by Chung et al, 8 suggests that the thieno-moiety in this and our MDCO molecules may contribute to apoA-I stimulation independent of bromodomain inhibition.
Footnotes
Author Contributions
Conceived and designed the experiments: DB, HK, SN, PF and OF. Analyzed the data: HK, SN, PF and OF. Wrote the first draft of the manuscript: HK. Contributed to the writing of the manuscript: PF, SN and SP. Agree with manuscript results and conclusions: PF, OF and SK. Jointly developed the structure and arguments for the paper: HK, PF and SK. Made critical revisions and approved final version: PF and SK. All authors reviewed and approved of the final manuscript.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.