
Abstract
Atherosclerosis, a disease characterized by plaque formation in the arterial wall that can lead to heart attack and stroke, is a principal cause of death in the world. Since the 1990's, protein nitrotyrosine formation has been known to occur in the atherosclerotic plaque. This potentially damaging reaction occurs as a result of tyrosine modification by reactive nitrogen species, such as nitrogen dioxide radical, which forms upon peroxynitrite decomposition or nitrite oxidation by hydrogen peroxide-activated peroxidase enzymes. The presence of protein-bound nitrotyrosine can be considered an indicator of a loss in the natural balance of oxidants and antioxidants, and as such, there is an emerging view that protein-bound nitrotyrosine may be a risk factor for cardiovascular disease. This review brings together evidence that the accumulation of protein nitrotyrosine during atherogenesis is more widespread than initially thought (as its presence can be detected not only in the lesion but also in the blood stream and other organs) and is closely linked to lipid intake.
Atherosclerosis
Atherosclerosis is a disease that represents one of the leading causes of death in the world. The pathogenic process involves a chronic inflammatory response by the arterial wall promoted by the migration of macrophages and build-up of low density lipoproteins (LDL) leading to plaque formation (Libby et al. 2002). Plaque rupture can lead to heart attacks and stroke. Extensive epidemiological research has identified four independent risk factors for atherosclerotic coronary heart disease (CHD) that include cigarette smoking (Auerbach et al. 1965), diabetes (Stamler et al. 1993), increased serum lipid and cholesterol content (Verschuren et al. 1995) and high blood pressure (Macmahon et al. 1990). Of these risk factors, there are several compelling aspects supporting the notion that cholesterol is the main villain (Roberts, 2006): (i) atherosclerosis can be reproduced in animals using a high fat, high cholesterol diet (Anitschkow, 1933) (ii) cholesterol is present in plaques (Leary, 1934), (iii) people with high serum cholesterol levels have a higher frequency of atherosclerotic events than those with lower serum levels (Kannel et al. 1971) and (iv) reducing serum levels of cholesterol and LDL cholesterol can decrease atherosclerotic events (Miettinen et al. 1972) and also diminish plaque size (Nissen et al. 2004). However, in a study that evaluated the four risk factors, it was found that amongst 87,869 male patients with CHD, 19.4% of this population lacked any of these conventional risk factors, and fewer than 1% presented with all four risk factors (Fig. 1) (Khot et al. 2003). These results indicate that while these conventional factors are useful predictors of disease in ~80% of the population, further research is necessary to understand the complex nature of atherosclerosis and coronary heart disease and to identify other risk factors.
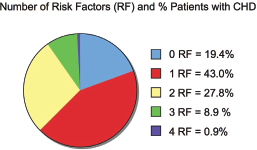
Other risk factors that are emerging include lipoprotein(a), fibrinogen, homocysteine, interleukin-8, interleukin-18 and high-sensitivity C-reactive protein (hs-CRP) (Danik et al. 2006; Boekholdt et al. 2004a; Ridker et al. 1999; Boekholdt et al. 2004b; Blankenberg et al. 2003; Ridker, 2008). In addition, there are now several reports of a link between protein 3-nitrotyrosine levels and the risk for cardiovascular disease and other conditions (Ohshima et al. 1990; Leeuwenburgh et al. 1997; Khan et al. 1998; Shishehbor et al. 2003; Troxler et al. 2004; Pennathur et al. 2004; Pirro et al. 2007; Hsiai et al. 2007). This review attempts to summarize the literature concerning the link between lipid, the inducible form of nitric oxide synthase (iNOS), 3-nitrotyrosine and atherosclerotic cardiovascular disease as well as potential steps that could be taken to eliminate protein nitration in this disease state.
Treatments for Atherosclerosis
Several treatments and new targets of therapy for atherosclerosis are known (described below), but given that there is emerging evidence that protein nitrotyrosine formation may be indicative of the oxidative and nitrative alterations that accompany atherosclerosis, very little attention to date has focused on reducing protein nitrotyrosine formation.
It is well known that lifestyle changes associated with diet, exercise and abstinence from smoking are associated with a lowered incidence of coronary heart disease (Stampfer et al. 2000). A mainstay in the treatment of cardiovascular disease is low-dose aspirin, which is thought to reduce the pro-thrombotic effects of thromboxane A2 (TxA2) produced by cyclooxygenase-1 (COX-1) in platelets (Ridker et al. 1991). A more recent, highly effective therapy in reducing the progression of atherosclerosis involves inhibitors of HMG-CoA (3-hydroxy-3-methylglutarylcoenzyme A) reductase, known as statins. Statins inhibit cholesterol biosynthesis and provide accelerated clearance by the liver of LDL that carries cholesterol as its main component. Several statins have been developed, e.g. atorvastatin, fluvastatin, simvastatin, pitavastatin, cerivastatin, rosuvastatin and pravastatin, all of which are structurally based on HMG-CoA and competitively inhibit the reductase (Illingworth and Tobert, 2001). Statins are among the most successful drugs developed in recent years with several clinical trials demonstrating their therapeutic benefit in the primary and secondary prevention of heart disease and are discussed in an excellent review elsewhere (Liao and Laufs, 2005). Despite their benefits in lowering cholesterol, statins have been described to have “pleiotropic” effects such as improving endothelial function, preventing plaque rupture and decreasing oxidative stress and inflammation (Liao and Laufs, 2005), as well as increasing HDL levels (McTaggart and Jones, 2008). Added evidence of the far-reaching effects of statins comes from the fact that the JUPITER trial (Justification for the Use of statins in Primary prevention: an Intervention Trial Evaluating Rosuvastatin) (Ridker et al. 2007), which evaluated the effect of 20 mg rosuvastatin among persons with average or low levels of LDL at increased cardiovascular risk due to enhanced levels of the inflammatory biomarker hs-CRP, was stopped prematurely (March, 2008) because of unequivocal evidence of a reduction in cardiovascular morbidity and mortality. Furthermore, as discussed below, the only therapeutic intervention studied to date for its effects on lowering 3-nitrotyrosine levels in patients with coronary artery disease or hypercholesterolemia involves statins (Shishehbor et al. 2003; Pirro et al. 2007).
The ACE (angiotensin-converting enzyme) inhibitors are usually used in the treatment of hypertension and congestive heart failure but have also been documented to reduce cardiovascular events (Yusuf et al. 2000b; Rueckschloss et al. 2002). The renin-angiotensin-aldosterone system is crucial in regulating blood pressure, but also plays a role in atherogenesis. In this regard, angiotensin II, which is produced from angiotensin I by ACE, is known to promote atherosclerosis in hyperlipidemic mice without changing blood pressure (Daugherty et al. 2000). ACE inhibitors have a beneficial effect on atherosclerotic disease in patients that do not exhibit high blood pressure but are at high risk for cardiovascular events (Yusuf et al. 2000b).
A major mechanism in the development of atherosclerosis is considered to be the overproduction of reactive oxygen species (ROS) that contribute to endothelial dysfunction in atherosclerosis (Yokoyama, 2004), however, clinical trials using antioxidants have yielded variable results. Although the American Heart Association recommends eating diets rich in antioxidants, the use of antioxidant supplements in suppressing cardiovascular disease has not always been borne out in clinical trials. For instance, treatment of patients at high risk for cardiovascular events with vitamin E (α-tocopherol) had no apparent effect on cardiovascular outcome (Yusuf et al. 2000a; Valagussa et al. 1999). On the other hand, more recent results from trials indicate a benefit in dietary intake of vitamin E with regard to reducing the incidence of chronic disease (Wright et al. 2006; Ascherio et al. 2005; Traber et al. 2008). For instance, the ASAP (the Antioxidant Supplementation in Atherosclerosis Prevention Study) trial, a 6-year study, demonstrated that combined slow-release vitamin C (250 mg) and E (136 IU) taken twice daily slowed atherosclerotic progression in hypercholesterolemic males (by 37%) (Salonen et al. 2003). The fact that divergent results have arisen from trials may result from: the derivative of tocopherol used, dosing regimen, a failure to measure levels of plasma antioxidant vitamins, biomarkers of inflammation and oxidative stress (Yokoyama, 2004). Thus, the negative evidence regarding vitamin supplements may be due to flaws in the design of the trials rather than proof of failure of vitamins in primary prevention (Traber et al. 2008). With regard to dosage, it is interesting to note that Linus Pauling, a two-time Nobel laureate, recommended taking 10,000 mg a day of vitamin C (Pauling, 1970). In his latter years, he purportedly took up to 18,000 mg per day of vitamin C, but succumbed to prostate cancer at 93 years of age. While vitamin C may have delayed the onset of his cancer by several years, it is difficult to verify since clinical trials to this date have not used such high doses.
It is only within the last 30 years or so, that an appreciation has developed for certain fats over others following a study of Greenland Eskimos who ingest fish oils but have a lower incidence of coronary atherosclerosis (Bang et al. 1976). Consumption of fish or fish oils containing the omega-3 fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) is associated with decreased cardiovascular death, whereas consumption of α-linolenic acid, a vegetable oil-derived omega-3 fatty acid, is less protective (Breslow, 2006). Mechanisms to explain how EPA and DHA are beneficial to cardiac health include the prevention of arrhythmias (Leaf et al. 2003), lowering of plasma triacylglycerides (Harris, 1997), decreasing blood pressure (Geleijnse et al. 2002), a reduction in platelet aggregation (Knapp, 1997), improved vascular reactivity (Harris et al. 1997), and suppression of inflammation (Calder, 2001). In contrast, the consumption of trans fatty acids has adverse effects, raising levels of LDL cholesterol, reducing levels of high-density lipoprotein (HDL) cholesterol and increasing the ratio of total cholesterol to HDL cholesterol, a powerful predictor of CHD (Mozaffarian et al. 2006).
Despite the well documented clinical trial results that certain lipoproteins and the renin-angiotensin-aldosterone system are important in the pathogenesis of atherosclerotic cardiovascular disease, new targets of therapy are currently under consideration and are highlighted in an excellent review elsewhere (Rader and Daugherty, 2008). The aforementioned review examines therapies that interfere with (i) lipoprotein metabolism and transport, (ii) intracellular signaling by biologically active lipids (e.g. blocking thromboxane, prostaglandin and leukotriene receptors and preventing lipid peroxidation), (iii) the ability of key adhesion molecules in the vascular wall to recruit and retain leukocytes, and (iv) the degradation of extracellular matrix components to prevent plaque rupture. However, none of the conventional treatments or new targets of therapy have focused on preventing protein nitrotyrosine formation, although accumulating evidence indicates that increased protein nitrotyrosine is correlated with the development of atherosclerosis.
Protein Nitrotyrosine Formation
Protein 3-nitrotyrosine formation involves a reaction between a tyrosine (Tyr) residue in a protein with a nitrogen oxide species and results in the addition of a -NO2 group to the 3- position of the tyrosyl phenol ring. The consequence of protein nitration can include alterations in primary and secondary structure, activity, and susceptibility of the protein to proteolysis.
Physiologically and pathophysiologically, the reactive nitrogen species responsible for protein Tyr nitration are likely derived from nitric oxide (NO) synthesized by a family of nitric oxide synthase (NOS) enzymes that include endothelial NOS (eNOS), neuronal NOS (nNOS) and iNOS. All NOS isoforms exist as dimers and require several cofactors including heme, flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), tetrahydrobiopterin (BH4), calmodulin and possibly zinc (Stuehr, 1997). During NOS activity, O2 and arginine are consumed to form NO radical as a by-product. Importantly, O2 consumption must be tightly coupled to electron transfer steps during the NOS catalytic cycle; otherwise the enzyme switches from producing NO to releasing superoxide anion (O2•-) (Xia et al. 1998; Vasquez-Vivar et al. 1998; Wever et al. 1997; Heinzel et al. 1992; Pou et al. 1999; Xia and Zweier, 1997).
Before the 1990's, NO was known only as a poisonous gas and toxic air pollutant. However, in 1998, R. Furchgott, L. Ignarro and F. Murad received the Nobel Prize “for their discoveries concerning NO as a signaling molecule in the cardiovascular system”. The seminal work of S. Moncada in the 90's also contributed greatly to our current understanding of NO, e.g. see (Knowles and Moncada, 1994). After this discovery, a paradigm shift occurred and the number of publications concerning NO increased from < 100 publications per year to 10–12,000 per year and is maintained at this level to this date. Physiologically, NO is involved in a multitude of reactions that are, for the most part, beneficial. For instance, an important function of NO is to dilate blood vessels (Furchgott and Jothianandan, 1991). Medications that operate on this principle include nitroglycerin, which relieves angina pectoris (chest pain from coronary artery disease) by releasing NO to relax blood vessels (Davis and Wiesel, 1955) and Viagra, which functions to prolong the vasodilating effects of NO (Boolell et al. 1996).
During many disease states, as well as during the aging process, ROS are formed (Ohara et al. 1993). Under such inflammatory, pathophysiological settings that include atherogenesis, iNOS is also broadly induced (Buttery et al. 1996). A potential problem is that NO, which is beneficial to the body, reacts with O2•- to produce the reactive nitrogen species peroxynitrite (ONOO–), which is a powerful oxidant. NO and O2•- combine at a near diffusion-limited rate (6.7 ± 0.9 × 109 l mol-1 s-1) and this out-competes the rate at which superoxide dismutase (SOD) scavenges O2•- (Huie and Padmaja, 1993; Beckman and Crow, 1993). Once formed, ONOO– may be involved in processes such as cell signaling (Upmacis et al. 2004) and enzyme activation (Landino et al. 1996; Upmacis et al. 1999), but is also involved in damaging reactions that include protein-bound nitrotyrosine formation (Fig. 2) (Beckman, 1996). Another pathway that may lead to protein nitrotyrosine accumulation involves nitrite (NO2-) metabolism by H2O2-activated myeloperoxidase (MPO) (Hazen et al. 1999), a major constituent of artery wall macrophages, as well as other peroxidase enzymes such as cyclooxygenase (COX; also known as prostaglandin H2 synthase, PGHS) (Schildknecht et al. 2005; Hajjar et al. 2006; Upmacis et al. 2006). An early citation of protein-bound tyrosine nitration goes back to the early 1900's and involves the nitration of fibroin, a protein created by silkworms in the production of silk (Johnson, 1915), although the earliest report of single amino acid tyrosine nitration is likely ascribed to Warren de la Rue in 1848, as described by Johnson et al. (Johnson and Kohmann, 1915). More recently, protein nitration has been associated with more than 50 disease states including atherosclerosis, Alzheimer's disease, cancer, acute lung injury and arthritis (Pacher et al. 2007). Protein nitration is thus, not only a potential indicator of inflammatory reactions that occur during atherosclerosis, but also of the oxidative and nitrative stress that occurs in other disease states.
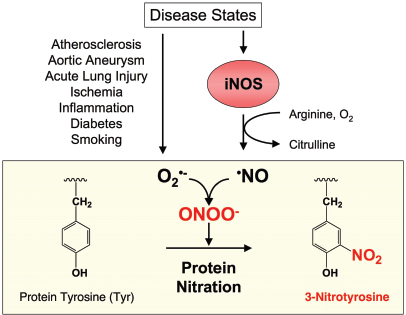
Protein Nitration in the Atherosclerotic Lesion
Beckman et al. first detected protein nitrotyrosine formation in early subintimal fatty streaks and in foamy macrophages within lesions of human coronary arteries in 1994 (Beckmann et al. 1994). The authors concluded that the presence of protein nitrotyrosine indicates that oxidants derived from NO are generated in human atherosclerosis and may be involved in its pathogenesis. Within the atherosclerotic lesion in mice and humans, we have demonstrated that COX is nitrated (Upmacis et al. 2001; Deeb et al. 2002; Deeb et al. 2006a; Deeb et al. 2006b). Furthermore, using mice missing the gene for ApoE (i.e. ApoE-/- mice; a murine model of atherosclerosis), we have shown that iNOS is the NOS isoform responsible for producing the NO-derived reactive nitrogen species responsible for protein nitration (Deeb et al. 2006b). COX enzymes initiate arachidonic acid metabolism in the biosynthesis of prostaglandins, but when a specific Tyr residue (Tyr385) crucial for catalysis is nitrated (by a heme-driven mechanism) COX inactivation results (Deeb et al. 2006a).
Additional targets of nitration in the atherosclerotic lesion have also been described. Notably, nitrated LDL in human lesions from the thoracic aorta was measured at levels 90 times higher (840 μmol/mol Tyr) than that in the plasma of healthy subjects (9 μmol/mol Tyr) (Leeuwenburgh et al. 1997). This result led to the proposal that higher oxides of NO may promote atherogenesis in opposition to the well-established anti-atherogenic effects of NO. Hsiai et al. determined that 5 specific Tyr residues are nitrated by ONOO–in the alpha and beta helices of apolipoprotein B-100 (apo-B-100) in LDL (Hsiai et al. 2007). Nitration of LDL is thought to interfere with cholesterol transport and trigger the release of TNF-α, which can amplify the inflammatory response (Smythe et al. 2003). Furthermore, nitrated LDL is absorbed by macrophages by the scavenger receptor pathway contributing to macrophage-driven foam cell deposition in the arterial wall (Graham et al. 1993).
Prostacyclin synthase (PGI2S), an enzyme with antithrombotic, antiproliferative, and vasodilatory functions in the normal vasculature, is also nitrated and inactivated in early stage atherosclerotic lesions (Zou et al. 1997; Zou et al. 1999; Schmidt et al. 2003). PGI2S nitration in atherosclerotic arteries prevents the rapid metabolism of PGH2 (the immediate product of COX metabolism and substrate for PGI2S), which serves as an agonist for the vasoconstrictive thromboxane receptor. Thus, overwhelming evidence exists that protein nitration occurs in the atherosclerotic lesion.
Protein Nitration at Sites Distant to the Atherosclerotic Lesion: Involvement of iNOS
Protein 3-Nitrotyrosine levels (pmol/mg protein) in ApoE-/- and ApoE-/-iNOS-/- mice fed either a regular chow or high fat and high cholesterol diet.
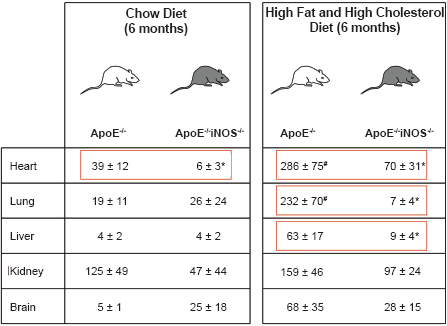
Adapted from Upmacis et al. (Upmacis et al. 2007).
P < 0.05 between ApoE-/- and ApoE-/-iNOS-/- mice on the same diet (within red box).
P < 0.05 between ApoE-/- mice on a high fat and high cholesterol diet and ApoE-/- mice on a chow diet.
Other data indicate that NO from iNOS is involved in atherosclerotic progression and plays a role in determining lesion size. In this regard, ApoE-/-iNOS-/-mice on a high fat, high cholesterol diet showed diminished atherosclerotic lesion formation compared to ApoE-/- mice (Detmers et al. 2000, Kuhlencordt et al. 2001). Thus, eliminating iNOS limits atherosclerosis, since NO-related species from iNOS are involved in the atherosclerotic progression.
Proteins that are known to be nitrated during cardiovascular disease are summarized in an excellent review (Peluffo and Radi, 2007). Targets of nitration in the vessel wall include apoB in LDL (Leeuwenburgh et al. 1997), COX (Deeb et al. 2002; Deeb et al. 2006b; Deeb et al. 2006a) and PGI2S (Zou et al. 1997; Zou et al. 1999; Schmidt et al. 2003), as discussed above. In addition, manganese superoxide dismutase (Mn-SOD) is readily inhibited by sequence-specific nitration at Tyr34 in the vessel wall upon aging (Yamakura et al. 1998; van der Loo et al. 2000) and also in rat kidneys during oxidative stress (Xu et al. 2006), and thus may also be a target of nitration during atherosclerosis. It is important to note that several proteins are also known to be nitrated in the plasma and in the myocardium. In the plasma, a 30% increase in nitrated fibrinogen has been detected in patients with documented coronary artery disease (CAD), which causes increased platelet aggregation and clotting (Vadseth et al. 2004). Nitration of plasmin interferes with its ability to hydrolyze fibrin fibers during clot resolution (Pignatelli et al. 2001; Nowak et al. 2004). Apolipoprotein A-I (ApoA-I), the major protein of HDL is also a target of nitration and results in a disruption in cholesterol transport (Zheng et al. 2004). In the myocardium, nitration of α-actinin leads to dysfunctional contractile processes (Borbely et al. 2005); nitration of the sarcoplasmic reticulum Ca2+ ATPase (SERCA) affects its activity and increased levels have been detected during human heart failure (Lokuta et al. 2005), as well as in aortae of atherosclerotic humans (Adachi et al. 2002; Xu et al. 2006); and nitration of myofibrillar creatine kinase (MM-CK) correlates with enzyme inactivation in humans with heart failure (Mihm et al. 2001). In summary, these findings support an emerging view that it would be important to measure protein 3-nitrotyrosine levels during atherosclerosis.
Protein 3-Nitrotyrosine Measurements in Humans
Summary of Protein Nitration Measurements in Disease.
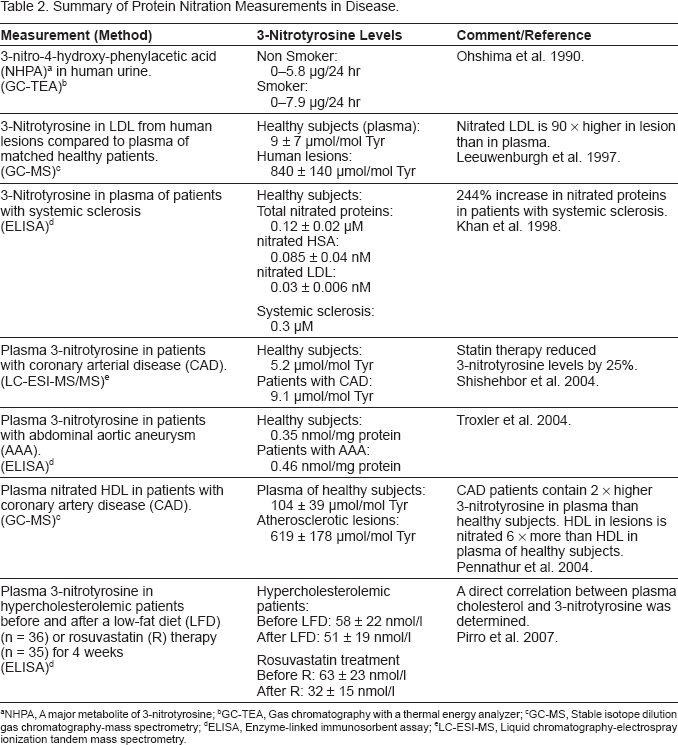
NHPA, A major metabolite of 3-nitrotyrosine
GC-TEA, Gas chromatography with a thermal energy analyzer
GC-MS, Stable isotope dilution gas chromatography-mass spectrometry
ELISA, Enzyme-linked immunosorbent assay
LC-ESI-MS, Liquid chromatography-electrospray ionization tandem mass spectrometry.
In 1997, using a highly sensitive method involving gas chromatography and mass spectrometry, Leeuwenburgh et al. determined that LDL isolated from the plasma of healthy subjects had very low levels of 3-nitrotyrosine (9 ± 7 μmol/mol of tyrosine) (Leeuwenburgh et al. 1997). In striking contrast, LDL isolated from atherosclerotic lesions had 90-fold higher levels of 3-nitrotyrosine (840 ± 140 μmol/mol of tyrosine). Thus, reactive nitrogen species, such as ONOO–, form in the human artery wall and promote LDL nitration in vivo.
In 1998, Khan et al. developed a semi-quantitative ELISA method to assay nitrated proteins in biological fluids and homogenates as a potential biomarker for oxidative stress (Khan et al. 1998). A concentration of 0.12 ± 0.01 μM nitro-BSA equivalents was measured in the proteins of normal plasma. Human serum albumin (HSA) and low-density lipoprotein (LDL) isolated from plasma contained 0.085 ± 0.04 and 0.03 ± 0.006 nmol nitro-BSA equivalents/mg protein, respectively. Thus, under normal conditions, nitration and oxidation of HSA and LDL are presumably inhibited by plasma antioxidants that may include ascorbic acid and thiols. In HSA of healthy subjects, this corresponds to one tyrosine nitrated per 3 × 107 tyrosines, i.e. one in 30 million Tyr residues. However, in patients with the inflammatory condition systemic sclerosis (an autoimmune connective tissue disorder) 0.3 μM nitro-BSA equivalents were detected. The authors also reported 2–100 fold larger levels of nitrotyrosine in lavages of patients with bronchial complaints (adult respiratory distress syndrome) than in lavages of normal patients.
In 2003, a study by Shishehbor et al. found that systemic levels of nitrotyrosine can be correlated with the prevalence of CAD and that statins can provide protection against these processes (Shishehbor et al. 2003). Nitrotyrosine levels were significantly higher among patients with CAD than healthy patient controls (median 9.1 μmol/mol Tyr vs. 5.2 μmol/mol Tyr; P < 0.001). Oral atorvastatin therapy (10 mg/day; 12 weeks) reduced nitrotyrosine levels significantly (25%; P < 0.02) with a magnitude similar to reductions in total cholesterol levels (25%; P < 0.001) and LDL particle number (29%; P < 0.001). The authors concluded that increased nitrotyrosine levels associated with CAD can be reversed by statin therapy. Thus, NO-derived oxidants play a role as inflammatory mediators in CAD and may have implications for atherosclerosis risk assessment.
In 2004, Troxler et al. noted that patients with aortic aneurysm have elevated levels of 3-nitrotyrosine (Troxler et al. 2004). Plasma 3-nitrotyrosine levels were measured in perioperative blood samples from patients undergoing repair of an abdominal aortic aneurysm (AAA) and from patients with intermittent claudication (poor circulation in the legs), smoking age-matched controls, non-smoking age-matched controls and non-smoking young healthy controls. In patients with an AAA, plasma 3-nitrotyrosine was significantly higher (0.46 nmol nitrated bovine serum albumin equivalents per mg protein) than levels measured in patients with intermittent claudication (0.35 nmol; P = 0.002), smoking controls (0–36 nmol; P = 0.001), non-smoking controls (0–35 nmol; P = 0.002) and young healthy controls (0.27 nmol; P < 0.001). Interestingly, AAA exclusion from the circulation reduced levels to control values (P = 0.001). Thus, the increased levels of circulating nitrated proteins in patients with an AAA compared with patients with claudication and controls suggests a greater degree of ongoing inflammation that was not related to smoking.
In 2004, Pennathur et al. reported that high density lipoprotein (HDL) in the circulation or artery wall is also damaged by reactive nitrogen species (Pennathur et al. 2004). The mean level of 3-nitrotyrosine in HDL isolated from human atherosclerotic lesions was 6-fold higher (619 ± 178 μmol/mol Tyr) than that in circulating HDL (104 ± 11 μmol/mol Tyr; p < 0.01). Their observations indicated that myeloperoxidase (MPO) promotes the formation of 3-chlorotyrosine as well as 3-nitrotyrosine in circulating HDL, but that other pathways, such as ONOO–, also produce 3-nitrotyrosine in atherosclerotic lesions. Levels of HDL isolated from plasma of patients with established CAD contained twice as much 3-nitrotyrosine as HDL from plasma of healthy subjects. These observations suggested to the authors that nitrated HDL might be a marker for clinically significant vascular disease and that reactive nitrogen species derived from nitric oxide promote atherogenesis. Thus, nitrated HDL may represent a previously unsuspected link between nitrative stress, atherosclerosis, and inflammation. As described earlier, further characterization of nitrated HDL reveals that the target of nitration is ApoA-I the primary constituent of HDL (Zheng et al. 2004). ApoA-I is associated with anti-atherogenic properties such as reverse cholesterol transport and anti-inflammatory properties. Nitration in HDL is associated with diminished ABCA1-dependent cholesterol efflux capacity of the lipoprotein and thus yields a dysfunctional form of HDL (Zheng et al. 2004). Indeed, a protective function of ApoA-I in diminishing levels of nitrative oxidants has been identified using ApoA-I-deficient mice, as levels of nitrated proteins in aortic tissues from these mice were 6-fold higher as compared to controls (Parastatidis et al. 2007).
In 2007, Pirro et al. investigated the short-term effect of cholesterol lowering with rosuvastatin on 3-nitrotyrosine levels, a marker of peroxynitrite-mediated oxidative stress, and on arterial stiffness (Pirro et al. 2007). They recruited 71 outpatients with primary hypercholesterolemia and assigned 35 patients to 4-week rosuvastatin therapy (10 mg daily) with a low-fat diet, and 36 patients to a low-fat diet only. The authors reported a positive correlation between cholesterol levels, 3-nitrotyrosine and aortic stiffness. Rosuvastatin therapy resulted in significant reductions in plasma cholesterol, 3-nitrotyrosine and aortic stiffness. Furthermore, reductions in both aortic stiffness and 3-nitrotyrosine levels correlated significantly with the decrease in plasma cholesterol. Thus, cholesterol reduction by short-term rosuvastatin therapy is associated with a decrease in peroxynitrite-mediated oxidative stress and improved large artery distensibility.
Removal of Nitrated Proteins from the System
Presently, it is unclear whether endogenous pathways remove nitrated proteins from the circulation, although it is possible that several mechanisms exist. There are now some indications that nitration may be a reversible process governed by a not yet fully characterized enzyme, known as a denitrase (Irie et al. 2003, Koeck et al. 2004). Identification of a denitrase would provide evidence that protein 3-nitrotyrosine is not formed randomly and has physiological significance in cellular signaling. There are also reports of proteases that target and degrade nitrated proteins at rates faster than native proteins (Souza et al. 2000, Gow et al. 1996). Immunoglobulins that recognize protein 3-nitrotyrosine and function to remove nitrated proteins from plasma could also play a role during inflammatory processes (Thomson et al. 2007). Indeed, specific immunoglobulins that are induced by Tyr-nitrated proteins have been found in the plasma of patients with acute lung injury. Thus, protein nitration is able to elicit an immune response, although it is not yet known whether the response is beneficial or harmful to the host (Thomson et al. 2007). Finally, ApoA-I may play a role in reducing nitrative burden, since removing this gene in mice causes an increase in nitrated fibrinogen levels (Parastatidis et al. 2007). Interestingly, induction of increased levels of circulating immunoglobulins that recognize 3-nitrotyrosine occurs concomitantly with increasing burden of nitrative oxidants (Parastatidis et al. 2007).
Specific Targeting of iNOS during Atherosclerosis
Given that iNOS-derived NO contributes to atherogenesis and that removal of iNOS has been shown to limit atherosclerosis in mice, an obvious therapy might involve the use of iNOS inhibitors. However, one needs to consider that despite its involvement in inflammation and in providing the main source of NO towards protein nitrotyrosine accumulation, iNOS also provides beneficial functions. For instance, iNOS plays a role in fighting infections and resisting tumor cell replication (MacMicking et al. 1995). With regard to infections, the lethal dose to Listeria monocytogenes is 10-times lower in iNOS-/- mice than wild-type mice (MacMicking et al. 1995). There is also evidence that iNOS partakes in wound healing, since wound closure in iNOS-/- mice is delayed by 31% compared to wild-type animals (Yamasaki et al. 1998). Thus, the trick would be to develop a drug to block the undesirable effects of iNOS without compromising the beneficial aspects. This remains a challenge in overall drug design by the pharmaceutical industry. An additional factor to consider is the intrinsic specificity of the drug designed to inhibit NOS. A non-specific inhibitor of NOS may lead to partial eNOS inhibition and a concomitant increase in blood pressure, as eNOS deletion in mice is known to lead to hypertension (Huang et al. 1995). Furthermore, inhibition can be complicated by the fact that deletion or inhibition of one isoform of NOS can lead to compensation by another form of NOS, e.g. see (Meng et al. 1998).
Despite concerns regarding the potential caveats of iNOS inhibition, clinical trials have provided limited information in terms of their usefulness. For instance, the TRIUMPH trial used a non-specific inhibitor of NOS, tilarginine acetate (also known as L-NMMA; produced by Arginox Pharmaceuticals), and tested the hypothesis that the inhibitor would reduce deaths associated with cardiogenic shock in patients with myocardial infarction (Hochman et al. 2007). Tilarginine produced a significant increase in blood pressure but provided no detectable protection against mortality versus placebo, leading to a discontinuation of this trial. Use of a non-specific NOS inhibitor in cardiogenic shock was not effective and will probably not be useful in other areas of cardiovascular disease. Topical application of an iNOS inhibitor (KD7040; produced by Kalypsys) is currently in phase IIa for the treatment of neuralgic pain that accompanies shingles. A phase II trial using another specific iNOS inhibitor (GW274150; produced by GlaxoSmithKline) in the treatment of acute migraine during the mild headache phase is also underway. Specific iNOS inhibitors have not been employed, however, in trials that are aimed at preventing atherosclerotic lesion development and reducing protein 3-nitrotyrosine accumulation. Despite its clear involvement in the progression of atherosclerosis and protein 3-nitrotyrosine formation, iNOS may prove to be a less than ideal target of therapeutic intervention.
Future Research
Studies indicate a link between a high fat and high cholesterol diet and protein 3-nitrotyrosine accumulation in organs of mice (Upmacis et al. 2007). Future research might involve an identification of those mechanisms that relate excessive lipid intake to increased protein 3-nitrotyrosine levels. Interestingly, results from clinical trials did not yield a significant correlation between 3-nitrotyrosine, LDL-C, HDL-C or total cholesterol, indicating that the tie between protein 3-nitrotyrosine production and lipid intake is likely independent of cholesterol (Shishehbor et al. 2003). Importantly, increased plasma protein 3-nitrotyrosine levels are associated with the presence of CAD (Shishehbor et al. 2003) and thus, a crucial step in preventing this disease state will be to gain an understanding of the pathways leading to its formation. While reports indicate that protein 3-nitrotyrosine accumulation during atherosclerosis is more widespread than initially thought, the full extent of the nature of all proteins modified is not yet known. Identifying these nitrated proteins/enzymes will allow us to make some conclusions about the mechanism and discover the true impact of these modifications on organ function. Furthermore, the exact locations of protein 3-nitrotyrosine formation within organs affected during atherosclerosis are not yet fully known. The identification of their location will allow us to conclude whether protein 3-nitrotyrosine formation is an extension of atherosclerosis (i.e. only associated with the blood vessel wall) or simply another process that accompanies atherosclerosis. Promising research has identified a major role for iNOS in the generation of reactive nitrogen species, particularly ONOO–, in protein 3-nitrotyrosine accumulation, and yet iNOS inhibition may not provide the most effective means of treatment. The statins currently provide the only therapeutic intervention studied to date in reducing 3-nitrotyrosine levels in patients, but their mechanism of action is not yet fully elucidated (Shishehbor et al. 2003; Pirro et al. 2007). Thus, unraveling the signaling pathways that occur upon excessive lipid intake that lead to iNOS induction and subsequent 3-nitrotyrosine production may hold the key to the development of future promising therapeutic interventions.
Footnotes
Acknowledgements
Supported, in part, by grants from the NIH (PO1 HL46403) and Philip Morris U.S.A. Inc., and Philip Morris International (to RKU), the Julia and Seymour Gross Foundation Inc., and an award from the Alice Bohmfalk Charitable Trust (to RKU).
The author reports no conflicts of interest.