
Abstract
The term “immune privilege” was originally coined to describe the suppression of inflammatory responses within organs protected by anatomic barriers, ie, the eyes, brain, placenta, and testes. However, cellular and metabolic processes, which orchestrate immune responses, also control inflammation within these sites. Our current understanding of tolerogenic mechanisms has extended the definition of immune privilege to include hair follicles, the colon, and cancer. By catabolizing tryptophan, cells expressing the enzyme indoleamine-2,3-dioxygenase produce kynurenine metabolites, which orchestrate local and systemic responses to control inflammation, thus maintaining immune privilege. This review highlights the double-edged role played by the kynurenine pathway (KP), which establishes and maintains immune-privileged sites while contributing to cancer immune escape. The identification of the underlying molecular drivers of the KP in immune-privileged sites and in cancer is essential for the development of novel therapies to treat autoimmunity and cancer and to improve transplantation outcomes.
Introduction
Immune privilege was first observed by van Dooremaal, a Dutch ophthalmologist who, in 1873, transplanted murine skin into the eyes of rabbits and dogs in an attempt to study the formation of cataracts.1,2 He was astonished by the observation that the grafted tissue was not rejected following its transplantation into the anterior chamber of the eye. In 1948, Medawar 3 reassessed the prolonged survival of foreign tissue grafted into the anterior chamber of the eye and coined the term “immune privilege”. He proposed that the apparent absence of lymphatic drainage protected the graft from immune rejection. In 1975, Kaplan et al. 4 showed that antigen introduced into the anterior chamber of the eye was able to induce a systemic downregulation of the antigen-specific cellular response, a process he called immune deviation. It is now also known that immunoregulatory processes contribute to immune privilege. In addition to the eye, other immune-privileged sites have been described and include the brain, placenta, and testes. More recently, it was discovered that similar immunoregulatory processes operate within the hair follicle and the colon; as a result, these organs were identified as immune-privileged sites despite lacking anatomic barriers. Furthermore, immune-privileged sites can be categorized based on their cellular regenerative capacity, with the eyes and brain exhibiting limited capacity, while the other sites contain stem cells that can repair damaged tissue.5,6
Novel findings emerging from systems biology studies involving metabolomics and transcriptomics have led to an expansion of the concept of immune privilege. 7 Based on the genome-wide transcriptome profiling of 23,843 genes, Doyle et al. 8 identified three key immune mechanisms that are involved in testicular immune privilege; these mechanisms include an immunosuppressive cytokine milieu, the presence of proteins regulating leukocyte trafficking through controlled cell junctions, and the inhibition of complement activation. Shechter et al. 9 recently proposed that the integrity of anatomic barriers within some immune-privileged sites is not absolute as shown by the existence of an epithelial “gate” beyond the endothelium vascular barrier in the testes; this gate allows selective cellular trafficking in response to inflammation. Furthermore, we and others have extended the concept of immune privilege to include chronically infected tissues and tissues in which cancer has escaped immune control; in both situations, immune privilege contributes to disease progression.10,11
In this review, we focus on the contribution of indoleamine 2,3-dioxygenase 1 (IDO1), and its immunosup-pressive catabolite kynurenine (Kyn), to immune privilege in different anatomic sites and in the setting of cancer. Upon activation, IDO1 catabolizes the essential amino acid tryptophan (Trp) into metabolites collectively known as kynurenines (Kyns) (Fig. 1).11,12 IDO-expressing myeloid cells locally exert a profound inhibitory effect on T cells, which in turn suppress the proinflammatory processes that occur in response to tissue damage, infection, and cancer. 13

A schematic representation of the key enzymes and metabolites in the kynurenine pathway.
IDO is at the center of the immune synapse, which bridges innate and adaptive immune responses involving antigen-presenting cells (APCs), such as dendritic cells (DCs) and macrophages (MPs), and lymphocytes, such as effector T (Teff) cells, natural killer (NK) cells, and regulatory T (Treg) cells.
The kynurenine pathway (KP) contributes to immune privilege within organs and does so in conjunction with the adenosine/purinergic pathway and with immune checkpoints, such as cytotoxic T lymphocyte antigen-4 (CTLA-4) and programmed cell death-1 (PD-1).14,15 The aim of this review is to highlight the double-edged role played by the KP, which beneficially contributes to maintaining immune-privileged sites to protect them from inflammation-mediated damage, while detrimentally participating in cancer immune escape.
The KP Plays a Critical Role in Immune Privilege
L-Trp is one of the nine essential amino acids required by humans and has a distinctive indole functional group. 12 Despite being the least abundant amino acid, Trp plays a considerable role in health and in disease given its roles in protein synthesis, serotonin production, and the KP (Fig. 1). The KP converts Trp into biologically active metabolites, including Kyn, kynurenic acid, 3-hydroxy Kyn, picolinic acid, quinolinic acid, and nicotinamide adenine dinucleotide (NAD). This process reduces Trp serum levels, which in turn decrease the availability of Trp, an amino acid required for microbial growth and cellular growth. These Trp metabolites and related enzymes have important and contrasting short-term antimicrobial and long-term immunosuppressive roles. 11 The Trp-catabolizing enzymes implicated in the KP are found in numerous cell types, including endothelial cells, fibroblasts, and myeloid cells.
The KP involves three distinct rate-limiting enzymes, such as IDO1 and IDO2, which are inducible enzymes in the gut mucosa and in other tissues, and tryptophan 2,3-dioxygenase (TDO), which is constitutively expressed in the liver and plays a dominant role in health. 16 TDO is also expressed at low levels in endothelial cells, neurons, astrocytes, and malignant cells, in conjunction with IDO1.17,18 TDO is upregulated by cellular stress, glucocorticoids, and Trp homeostasis. Conversely, IDO1 can be dramatically enhanced during inflammation, making it the most important of the three enzymes during inflammatory diseases. 19 IDO2, which is not redundant with respect to IDO1 function, is present at a lower level than IDO1 and is expressed by similar cells as IDO1. 20 In this review, the term IDO refers to IDO1 unless specified otherwise.
A variety of cells express IDO, including monocytes, MPs, DCs, endothelial cells, fibroblasts, and certain cancer cells.21,22 IDO is upregulated by inflammatory molecules, such as amyloid peptides and lipopolysaccharides, and by inflammatory cytokines, such as interleukin-1 (IL-1), interferon gamma (IFN-γ), and tumor necrosis factor alpha (TNF-α).23,24
IDO-deficient mice and mice treated with IDO inhibitors fail to tolerate the fetus during allogeneic pregnancy, 25 experience colitis due to the failure of mucosal tolerance of the intestinal microbiota, 26 and lose the ability to clear apoptotic cells. 27 Furthermore, blocking or ablating IDO expression worsens inflammation in models of graft-versus-host disease, 28 autoimmunity, 29 and chronic conditions, such as chronic granulomatous disease and diabetes,30,31 illustrating the key role played by IDO in controlling inflammation. In all these models, IDO inhibition can represent an interesting target as it is induced by inflammation, while inhibition of CTLA-4 may be clinically beneficial at the expense of developing autoimmune conditions as its expression is constitutive in Treg cells and induced in conventional T cells.13,32
The KP contributes to immune privilege by the following four mechanisms: (1) by depleting Trp via the induction of the stress response kinase, general control nondepressible 2 (GCN2), and the suppression of mammalian target of rapamycin 1 (mTOR1) pathway, which senses amino acid withdrawal, a process inhibiting Teff cell function and growth (Fig. 2)33,34; (2) by the direct effect of Kyn on the aryl hydrocarbon receptor (AhR), which induces DC and MP differentiation; these cells in turn first induce inflammatory T17 cells followed by a global reprogramming driving their conversion (transdifferentiation) to Treg cells upon the resolution of inflammation. 35 In the context of chronic inflammation, the effect of Kyn on AhR is directed toward a predominant immune suppression; (3) by promoting the differentiation of CD4 T cells into Treg cells expressing CTLA-4 and via phosphatase and tensin homolog, a protein encoded by a tumor suppressor gene, which is mutated in several cancers 36 ; and finally (4) by the Kyn-mediated inhibition of IL-2 signaling, which impairs memory CD4 T cell survival.37–39 Upon IDO activation, APCs capable of producing inflammatory cytokines such as IL-12 switch to produce inhibitory cytokines such as transforming growth factor beta (TGF-β) and IL–10. 37 In addition, IDO can be triggered in the presence of apoptotic cells to induce self-tolerance and/or cancer persistence. 40 Globally, IDO activation can transform the function of APCs and convert local T cell function from an immunogenic one to a tolerogenic one. The relative contribution of the KP to the various organs characterized by immune privilege will be described in the following sections.

The role of the kynurenine pathway in inducing tolerance in immune-privileged sites, in tumors, and in the tumor microenvironment.
The KP as a Guardian of the Eyes and Brain
The eye and the central nervous system (CNS) are formed from the neuroepithelium during embryonic development, and they each have a nonexpandable anatomic barrier. 9 Since these barriers do not permit swelling in the case of inflammation and these sites have limited cellular regenerative capacity, immune privilege is a necessary protective feature.
The eyes
The concept of immune privilege originated from experimental animal models in which allografts were accepted by the eyes but rejected by the skin.41,42 The eye is anatomically divided into the following three layers: the outer or fibrous layer (the cornea/sclera), the middle or vascular layer (the uvea-iris, ciliary body, and choroid), and the inner or sensorineural layer containing the blood–retina barrier. The blood–retina barrier contributes to immune privilege in the eye.
Over the last decade, the contribution of the IDO expressed by the human corneal cells lining the anterior chamber of the eye to ocular immune privilege has gained considerable attention.2,43,44 IDO is expressed in many ocular tissues and contributes to the immune privilege of corneal allografts. 2 In vitro studies have shown that T cells cocultured with iris and ciliary body cells acquire Treg activity induced by IDO and TGF-β expression.40,45
Other ocular tissues such as the uvea, the conjunctiva, and the periocular fascia contain IDO-expressing MPs and tolerogenic DCs secreting immunosuppressive cytokines; these cytokines inhibit Teff cells and induce Treg cells. 41 Other mechanisms involved in inducing immune privilege within the eye include programmed cell death ligand 1 (PD-L1), macrophage migration inhibitory factor (MIF), TNF-related apoptosis-inducing ligand (TRAIL), and Fas ligand (FasL), all of which also inhibit T cell proliferation and survival and increase Treg cell numbers (Table 1), which was recently reviewed by Niederkorn et al. 2 and Benhar et al. 46 In addition, NK and γδ T cells were found to contribute to immune privilege in the murine eye, as did the reduced expression of MHC molecules.47–50
A summary of selected studies which have examined the contributions of the kynurenine pathway and other factors to immune-privileged sites and to cancer and its microenvironment.

The brain
In 1913, Goldmann 51 was the first to report the existence of a blood-brain barrier (BBB) using dyes devised by Ehrlich. 52 Goldmann observed that systemically injected trypan blue dye failed to penetrate the brains of animals as readily as it penetrated other tissues. Although, the term “BBB” was first used as early as 1900 by Ehrlich's student Lewandowski, it was not until the 1960s that the actual existence of the BBB was proven by the elucidation of its cellular structure.53,54
Several types of neural cells are known to produce KP metabolites along with varying levels of KP enzymes. In 2001, Guillemin et al. 55 were the first to report that astrocytes produced significant quantities of Kyn, which, by inducing T cell suppression, plays a protective role against neuronal excitotoxic-induced cell death.56–58 Kwidzinski et al. 59 showed that microglial cells, which are able to inhibit T cell responses by both Trp depletion and Treg cell induction, increase IDO expression upon IFN-γ stimulation in vitro and in vivo. Using an experimental murine autoimmune encephalomyelitis (EAE) model, which is the result of a breach in immune tolerance to CNS antigens and resembles multiple sclerosis in humans, Kwidzinski et al. 60 also showed that the administration of the IDO inhibitor 1-methyl-D-tryptophan exacerbated this condition. Similarly, Yan et al. 29 have shown that in IDO-deficient mice, enhanced inflammatory T1 and T17 responses exacerbated EAE. Conversely, the administration of 3-hydroxyanthranilic acid, another downstream KP metabolite, was associated with an increased proportion of Treg cells and with an inhibition of the T1/T17 response, which reduced EAE severity.
In multiple sclerosis patients, IDO gene expression and activity are predictive of relapse. 61 These findings indicate that during inflammation, the KP orchestrates self-protective mechanisms, limiting antigen-specific immune responses in the CNS. Other factors implicated in brain immune privilege have been recently reviewed by Louveau et al. 62 and are summarized in Table 1.
Maternal Acceptance of the Fetus: Transgenerational Tolerance
In a pregnant woman, the fetus is a semihaplotypic allograft that is nonetheless tolerated by the maternal immune system. 63 As initially reported in 1961 by Medawar, 64 the immune tolerance exerted by the placenta protects the fetus from maternal immune responses directed toward paternally inherited antigens. In rare cases, spontaneous abortions occur due to a break in immune tolerance induced by placental inflammation.
The maternal vasculature does not penetrate the fetal parenchyma, while the placenta serves as the fetomaternal barrier. 65 The contribution of the KP to maternal immune tolerance of the fetus was discovered in 1998 by Munn et al. 25 It had been previously shown that the complement system also contributes to fetal tolerance. 66 The placenta is not considered as a strict anatomic barrier since the inhibition of Trp metabolism was observed to induce potent fetal T cell responses in mice, indicating that the KP contributes significantly to immune tolerance of the fetus. In 2004, Aluvihare et al. 67 showed that IDO-myeloid dependent induced Treg cells contributed to maternal tolerance of the fetus. Moreover, IDO expression has been observed in extravillous trophoblasts and in chorionic villi within the placenta. 68 Importantly, Zenclussen et al. 69 showed that the adoptive transfer of pregnancy-induced Treg cells prevented fetal rejection in a murine abortion model. In humans, Somerset et al. 70 showed that normal human pregnancy is associated with an increased number of immunosuppressive Treg cells, thus confirming the observations made in the murine model.
Other factors associated with immune privilege within the gravid uterus include the expression of inhibitory PD-L1 and CTLA-4 by both Treg cells and trophoblast giant cells, the expression of MIF by trophoblasts, the inhibition of NK cell cytotoxicity, and the upregulation of apoptosis-inducing FasL expression in the human endometrium (Table 1).63,68,71 Furthermore, in the pregnant uterus, MPs with an anti-inflammatory M2 phenotype secrete immunosuppressive IL-10 and prostaglandin E2, in addition to IDO expression.72,73 Moreover, the hormonal control of the innate and adaptive immune responses plays a particularly important immunosuppressive role, as has been previously reviewed by Wira et al. 74 It has also been shown that the placental expression of the nonclassical MHC class I E, F, and G molecules, which can activate certain inhibitory receptors expressed on cells of lymphoid and myeloid origin, contributes to maternal–fetal tolerance. 75
The KP Contributes to Testicular Immune Privilege
The testes are an immune-privileged site that tolerate new germ cell autoantigens generated during puberty, a period of immune competence (Fig. 3). 76
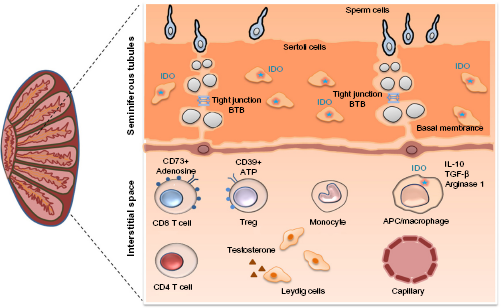
The kynurenine pathway enzyme IDO plays a central role in testicular immune privilege.
Similarly to the eyes and CNS, testicular immune privilege was initially attributed to the presence of a barrier, specifically the blood–testis barrier (BTB). 76 The BTB limits the passage of antibodies into the testes, in addition to limiting the access of germ cell antigens to testicular interstitial immune cells. In 1980, Yoshida et al. 77 reported that rodent epididymal and testicular extracts had high IDO activity. Later, Hansen et al. 78 demonstrated that, unlike the inflammation-induced expression of IDO that occurs in nearly all mammalian tissues, the expression of IDO in the testes is constitutive. Testicular immune privilege has been extensively characterized in rodents in which attenuated innate and adaptive immune responses have been shown to be associated with reduced T cell numbers, except for IDO-myeloid-induced Treg cells whose numbers are increased, and with the enhanced production of several anti-inflammatory cytokines including IL-10 and TGF-β.76,79,80
Sertoli cells (SCs) are found in the testes, where their main function is to provide local immune tolerance and nourishment to developing germ cells. SCs were used as a therapeutic strategy in autoimmune diabetes to self-protect and coprotect allogeneic and xenogeneic pancreatic grafts from immune destruction in different experimental settings. In a murine cotransplantation model, Fallarino et al. 81 showed that inhibiting the IDO activity abrogates the ability of porcine SCs to protect pancreatic islet allografts. Furthermore, efficient Trp metabolism was linked to the protective effects of SCs, an effect that involved the IDO- and TGF-β-dependent emergence of autoantigen-specific Treg cells.
SCs, also called “mother cells”, provide myeloid and lymphoid immune tolerances to protect germ cells in the testes and cotransplanted cells at ectopic sites. This is further supported by Jrad-Lamine et al. 82 who showed that the level of IDO expression in the murine epididymis is higher compared to that in tissues in which IDO expression is induced.
Understanding human testicular immune privilege in health and in disease has been limited by the difficulty of sampling testicular tissue. Recently, we were the first to assess the complex immunological milieu of the human testes in healthy and in human immunodeficiency virus (HIV)-infected adults who had elected to undergo gender reassignment surgery. 83 Healthy testes expressed significantly higher levels of IDO compared to blood, as determined by quantitative PCR. Interestingly, we also observed the contribution of another immunometabolic pathway, the adenosine–adenosine triphosphate (ATP) pathway, to testicular immune privilege. This signaling pathway is characterized by its location on the extracellular surface of the cell membrane. In contrast to ATP, which acts on immune cells to promote inflammation, its metabolite adenosine functions as an anti-inflammatory molecule. The extracellular ectoenzymes CD39 and CD73, which are expressed mainly on T cells, dephosphorylate extracellular ATP, generating adenosine monophosphate (AMP) and immunosuppressive adenosine, which induces immune tolerance by blocking IL-2 signaling.84,85 Of interest, we observed significantly higher frequencies of adenosine-dependent, immunosuppressive CD39+ Treg cells and CD73+ memory CD8 T cells in the testes than in the blood. We also observed that both the KP and adenosine-ATP immunosuppressive pathway are involved in testicular immune privilege. 83
Other factors that contribute to testicular immune privilege include the secretion of immunosuppressive IL-10 and TGF-β by MPs, and by peritubular, Sertoli, and testosterone-secreting Leydig cells,76,79,80 which was recently reviewed by Cutolo 86 and Li et al. 87 (Table 1).
The Hair Follicle and Colon: Immune-Privileged Sites without Anatomic Barriers
In 2005, Paus et al described immune privilege-like mechanisms in hair follicles; these mechanisms included IDO expression by tolerogenic APCs, low-level expression of MHC class I, and high-level expression of TGF-β and IL-10.88–90 Hair follicles are, therefore, organs with distinctive immunologic characteristics, which was recently reviewed by Paus and Bertolini. 89 Alopecia areata, an autoimmune disease resulting from the loss of immune privilege within the hair follicles, is characterized by T cell cytotoxicity toward the follicles, which causes transient, nonscarring hair loss. 91 Encouragingly, the use of Ruxolitinib, a JAK2 inhibitor, blocks CD8 T cell cytotoxic function, leading to an improvement of alopecia.
Another organ without anatomic barriers is the intestinal mucosa, which encompasses epithelial cells, the lamina propria, and the mucosal immune system within the gut. 92 This organ, which has the largest surface area of all of the organs, separates the human body from the outside world. Since the intestine must balance the need to tolerate food and commensal flora while controlling invading pathogens, the intestine can be considered to be an unconventional immune-privileged site. The KP was recently shown to play a significant role in establishing intestinal immune privilege. In 2013, Zelante et al. 93 showed that Trp catabolites generated by certain Lactobacilli species contribute to mucosal tolerance of the commensal flora, known as the microbiota. Catabolites such as Kyns, which are AhR ligands, promote local IL-22 production by innate lymphoid cells type 2, resulting in protective intestinal homeostasis. Furthermore, Vujkovic-Cvijin et al. 94 recently reported that the Lactobacilli present in stool were associated with IDO upregulation, which contributed to the tolerance of the microbiota by the gut mucosa in simian immunodeficiency virus (SIV)-infected macaques.
Recently, Lamas et al. 95 determined that mice lacking the CARD9 gene were more susceptible to colitis due to the defective activation of the gene for IL-22, a cytokine involved in gut homeostasis and repair. Furthermore, Kyn generated by the catabolism of Trp by Lactobacillus increased the production of IL-22 via ligation to AhR. To establish the causal role of the microbiota in colitis, CARD9−/− germ-free mice were supplemented with Lactobacillus strains capable of metabolizing Trp; this restored IL-22 expression and AhR ligand production, which decreased susceptibility to colitis. Stool samples from patients having inflammatory bowel syndrome (IBD) showed a reduced activation of AhR compared to those form healthy controls. These results illustrate the important link between host inflammatory genes, IBD, Trp metabolites, and Lactobacilli, with the production of AhR agonists in colon homeostasis.
Other factors involved in intestinal immune privilege have been reported and include FasL, TRAIL, thymic stromal lymphopoietin, prostaglandin E2, retinoic acid, TGF-β, and IL-10 (Table 1).96–98 These substances contribute to the induction of tolerogenic DCs. Treg cells also contribute to immune privilege as reported by Coquerelle et al. 99 who showed that in a murine colitis model, administration of anti-CTLA-4 monoclonal antibody induced the development and/or expansion of IL-4- and IL-10-producing Treg cells in an IDO-dependent manner, which improved the colitis.
Overall, the findings of these various studies involving different animal models highlight the therapeutic potential of KP-associated immune privilege.
Cancer and its Microenvironment: Kyn is the Enemy Within
In 2008, Mellor and Munn 10 extended the definition of immune privilege to include cancer and its microenvironment. In the early stage of tumor development, proteins encoded by mutated genes are thought to be the major source of antigens in cancer cells, which are able to circumvent tumor-specific immune responses owing to the immune-privileged tumor or its microenvironment.15,100,101
IDO can be expressed by tumor cells and by myeloid cells surrounding the tumor; these myeloid cells include APCs, tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs), which mediate an acquired immune tolerance toward the tumor, thereby thwarting host immune responses. It was recently recognized that the tumor's metabolic microenvironment plays a major role in maintaining immune responses by causing a shift from mitochondrial oxidative phosphorylation, an efficient way to generate ATP, to a less efficient aerobic glycolysis pathway in cytotoxic T cells (CTLs). 102 This metabolic reprogramming or Warburg effect within the tumor and its microenvironment leads initially to a local immune exhaustion involving CTLs and NK cells, which will become systemic when metastasis develops. Such cancer-related immune exhaustion has recently been shown to be influenced by the gut microbiota in a distal nondigestive tumor. 103 Furthermore, cytokines and chemokines secreted by the hypoxic and necrotic zones of the tumor microenvironment attract TAMs; the presence of high numbers of TAMs expressing high levels of IDO and arginase-1 is a phenomenon linked to immune escape and to faster disease progression.
The tumor microenvironment also accumulates immature and tolerogenic tumor-infiltrating APCs expressing low levels of costimulatory proteins such as CD80 and CD86; thus, these APCs lack the ability to stimulate naive T cells. 102 The migration of these APCs into the draining lymph nodes in which the APCs present tumor antigens to tumor-specific T cells subsequently results in their failure to activate T cells. In mice, MDSCs infiltrate the tumor microenvironment and promote T cell dysfunction and Treg cell generation through the expression of IDO and arginase- 1. 104 Targeting MDSCs with monoclonal antibodies has been shown to restore the tumoricidal function of tumor-infiltrating T lymphocytes in mice.
In 2003, Uyttenhove et al. 105 were the first to describe the constitutive expression of IDO by either the tumor cells or the APCs in the tumor microenvironment of various human cancers including cancers of the lung, pancreas, stomach, prostate, endometrium, and ovaries. They also showed that the expression of IDO by immunogenic murine tumor cells prevented their rejection by preimmunized mice, thereby inducing immune privilege. This effect was associated with the inhibition of specific T cell responses at the tumor site, an effect that was partly reversed by the administration of an IDO inhibitor. Subsequently, a number of studies reported an important role for the KP in several other cancers.106–109
Another immunometabolic pathway involving adenosine and ATP was recently described.85,110 Pericellular adenosine in the tumor microenvironment suppresses antitumor T cells by binding to the adenosine A2 A receptor, which activates cyclic adenosine monophosphate/protein kinase A signaling; this pathway subsequently inhibits IL-2 production in T cells, thereby suppressing the function of Teff cells. Interestingly, we have recently shown in vitro that a physiological concentration of Kyn (5 μM) was sufficient to reduce the ability of memory CD4 T cells to respond to IL-2, 39 therefore identifying a mechanism that can be therapeutically targeted.
Other factors contributing to immune privilege within tumors include the following immune checkpoints and immunosuppressive cytokines: (1) immune checkpoints like PD-L1 and/or PD-L2, CTLA-4, and Fas, which are expressed at higher levels in the tumor and its microenvironment are the targets of several immune interventions; and (2) the influence of cytokines which was first reported in 1989 by Werner et al. 111 who showed that the influence of IFN-γ on IDO expression in cell culture using various human cells. Litzenburger et al. 112 established a link between AhR and the IL-6/STAT3 axis in driving IDO expression. However, Li et al. 113 have shown that the long-term maintenance of IDO expression was found to be independent of IFN-γ, IL-10, TGF-β, TNF-α, and IL-6, contrasting with IDO enzymatic activity and IFN-γ-induced AhR expression. These immune checkpoint molecules and cytokines have been recently reviewed by Galluzzi and Kroemer, 114 and their inhibition by therapeutic antibodies is under intense clinical evaluation (Table 1).
KP-Associated Immune Privilege as an Immunotherapeutic Target
Given the dual role of the KP in immune tolerance and in cancer immune escape, this pathway is being investigated as an immunomodulator in autoimmune conditions and in cancer. Encouragingly, due to its upstream effect on the antigen-presenting milieu, IDO is nonredundant with more distal T cell checkpoints. Thus, blocking IDO concurrently with CTLA-4/PD-1 might confer additive benefits.
Recently, pharmacological, genetic, and immunological methods targeting IDO in a number of animal tumor models have shown therapeutic benefits. 115 Furthermore, several pharmacological IDO inhibitors are currently undergoing clinical evaluation.116–118 In particular, indoximod, a competitive inhibitor of IDO, was shown in a Phase I/II study to induce tumor responses in individuals with metastatic solid tumors.118,119 A Phase 1, dose-escalation study of epacadostat (INCB024360) in patients with advanced solid tumors has been initiated to assess the tolerability and efficacy of this novel IDO inhibitor, which is a specific IDO1 enzymatic inhibitor, versus indoximod, which acts through multiple targets within the KP. 120 The ongoing clinical studies of IDO inhibitors have been recently summarized.115,118
Finally, studies of combination therapy conducted by Wainwright et al. 121 showed that simultaneously blocking the expression of IDO, CTLA-4, and PD-L1 in a murine model of malignant glioma, a brain tumor with poor prognosis, provided significant therapeutic benefits. These encouraging preclinical results suggest that this combinatorial immunosuppressive strategy should be assessed in patients with glioma. Furthermore, while combining antibodies to CTLA-4 and PD-1 induced cumulative toxicities in human trials, preliminary data in patients with metastatic melanoma indicate that combining IDO inhibitors with either CTLA-4 inhibitor or PD-1 inhibitor is very effective and does not cause additive immune toxicity.122,123
Preclinical studies of IDO2 and TDO inhibitors as cancer treatments are also ongoing.118,124 The results of these studies are expected to provide novel insights into the contribution of the KP to immune-privileged sites and to cancer.
Conclusion
The KP has unique features distinguishing it from those of other immunosuppressive processes that are also involved in inducing immune privilege. The KP, which affects cell growth and proliferation, functions in conjunction with immunometabolic processes that induce tolerance by bridging myeloid and lymphoid cellular interactions. IDO, the major KP enzyme, has emerged as a key player in the establishment and maintenance of immune privilege within the eye, brain, placenta, and testes, and also appears to be involved in tumor immune escape. It is expected that current and future studies investigating immunotherapeutic approaches that control the activity of KP enzymes such as IDO and TDO, and of key metabolites such as Kyn and 3-hydroxykynurenine, will lead to the development of new treatments, which will improve the clinical outcomes of transplant, autoimmune, and cancer patients.
Author Contributions
Conceived and designed the review: J-PR, VM. Wrote the first draft of the article: J-PR, VM. Contributed to the writing of the article: J-PR, BR, GMG, VM. Jointly developed the structure and arguments for the article and contributed to the figures and table: VM, BR, GMG, J-PR. Made the critical revisions and approved the final version: J-PR, GMG. All authors reviewed and approved the final article.
Footnotes
Acknowledgment
The authors are grateful to Angie Massicotte for administrative assistance.