
Abstract
Monoclonal antibodies (MAbs) are an old immunological tool with applications in the fields of immunology, biotechnology, biochemistry, and applied biology. Production of monoclonal antibodies using hybridoma technology was discovered in 1975 by Georges Kohler of West Germany and Cesar Milstein of Argentina. Modern-day research on MAbs from laboratories worldwide is revealing additional applications in diverse branches of sciences. Recently, MAbs have been widely applied in the field of clinical medicine. Currently, MAbs account for one-third of all the new therapeutic treatments for breast cancer, leukemia, arthritis, transplant rejection, asthma, and psoriasis, with many more late-stage clinical trials being conducted. In this review, we outline the (i) production of MAbs, (ii) application of MAbs, (iii) antibody engineering, and (iv) pharmaceutical application of MAbs. The future prospect of this review lies in the applicability of monoclonal antibodies as a molecule for understanding and monitoring the biology of disease and its role in research, clinical, diagnostic, analytical, and pharmaceutical applications.
Introduction
The immune system in vertebrates is continuously evolving to protect itself from different intruding pathogens. The immune responses rotate around some innate mechanisms, including adaptive processes such as producing antibody (Ab) molecules that can bind to all molecular structures of the microbial pathogen (bacteria, viruses, fungi, nematodes, and other parasites) and can keep pace with the diversified mutations in anorganism. An antigen is defined as a molecule or part of a molecule that can be recognized by the immune system as a foreign entity. The challenge of the immune system is thus combated in two ways. First, through an antibody diversity mechanism, B lymphocytes produce varied antibodies specific for a new antigen (epitope) expressed by a pathogen by shuffling and reshuffling its genetic constituents. Second, paratope-encoding genes of the antibody are mutated rapidly to cope and bind strongly with the epitope of the antigen. Thus, these generated antibodies are better at binding with the antigen with greater affinity and high specificity. 1
Therefore, antibodies are useful research tools in diagnosis and therapy, as they can recognize and bind specifically and strongly with respective antigens. Polyclonal antibodies mixtures contain different antibodies developed in the blood of immunized animals from different cell types. As most antigens bear multiple epitopes, they can stimulate the proliferation and differentiation of a variety of B-cell clones. Thus a heterogeneous pool of serum antibodies can be produced with specificity for particular epitope(s) of the antigen.
In contrast, monoclonal antibodies (MAb(s)) are a mixture of homogenous antibody molecules with affinity towards a specific antigen, often generated using a hybridoma by fusing a B-cell with a single lineage of cells containing a definite antibody gene. Finally, a population of identical cells (or clones) is produced that secrete the same antibody.
Due to their specificity and high reproducibility using culture techniques, MAbs offer advantage over polyclonal antibodies. MAbs are increasingly used in applications such as research and diagnosis, therapeutic tools in cancer and immunological disorders, and pharmacy, thus generating a great demand in industry. The essential characteristics that confer the clinical applicability of MAbs include their specificity of binding and homogeneity, as well as their ability to be produced in unlimited quantities. 2 Another unique advantage of hybridoma production is that a mixture of antigens can be employed to generate specific antibodies. This also enables one to screen an antibody of choice from a mixture of antibody population with a purified antigen; thus, a single cell clone can be isolated.1–2 For these reasons, the objective of this review was to dissect the diverse facets of applicability of MAbs in disease monitoring and diagnosis.
History
The production of MAbs by hybridoma technology was discovered in 1975 by Georges Kohler of West Germany and Cesar Milstein of Argentina, who jointly with Niels Kaj Jerne of Denmark were awarded the Nobel Prize for Physiology and Medicine in 1984. In 1976, Kohler and Milstein developed a technique to fuse splenocyte cells (separated from the spleen of an immunized mouse) with tumorous myeloma cells. The hybrid cells were clones of antibody producing cells against a desired antigen and propagate rapidly to produce very large amounts of antibody. The hybridoma is capable of rapid propagation and high antibody secreting rates such as in myeloma cells, which can maintain the antibody genes of mouse spleen cells. In 1988, Greg Winter used the first humanized MAbs to avoid reactions/ responses observed in patients injected with murine derived MAbs.1–4
Outline of Production of MAbs
The main objective is to produce a homogenous population of MAbs against a pre-fixed immunogen (Fig. 1). 2 The basic strategy includes (i) purification and characterization of the desired antigen in adequate quantity, (ii) immunization of mice with the purified antigen, (iii) culture of myeloma cells which are unable to synthesize hypoxanthine-guanine-phosphoribosyl transferase (HGPRT) enzyme necessary for the salvage pathway of nucleic acids, (iv) removal of spleen cells from mice and its fusion with the myeloma cells, (v) following fusion, the hybridomas were grown in hypoxanthine aminopterin thymidine (HAT) medium. The fused cells are not affected in the absence of HGPRT unless their de novo synthesis pathway is also disrupted. In the presence of aminopterin, the cells are unable to use the de novo pathway and thus these cells become auxotrophic for nucleic acids as a supplement to HAT medium. In this medium, only fused cells will grow. Unfused myeloma cell does not have ability to grow in this HAT medium because they lack HGPRT, and thus cannot produce DNA. Unfused spleen cells cannot grow because of their short life spans. Only fused hybrid cells or hybridomas can grow in HAT medium. Hybrid cells have the capacity to grow in the HAT medium since spleen cell partners produce HGPRT. (vi) The hybrid cell clones are generated from single host cells (vii) the antibodies secreted by the different clones are then tested for their ability to bind to the antigen using an enzyme-linked immunosorbent assay (ELISA). (viii) The clone is then selected for future use.1–2
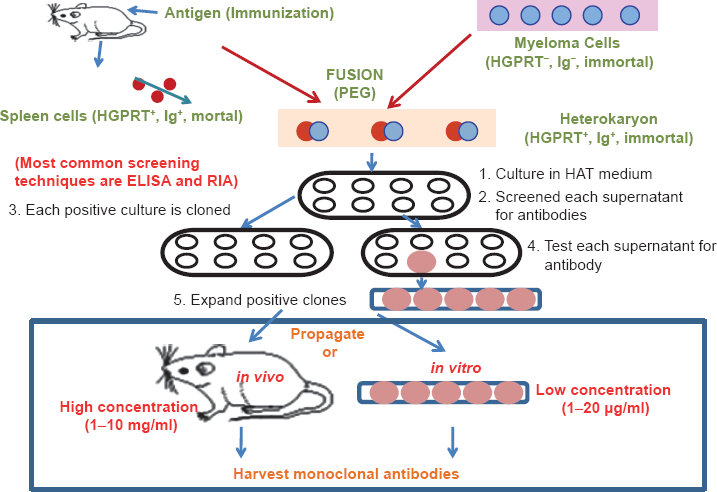
Immunization schedule
Depending on the purity and nature of the purified antigen, an immunization protocol is determined. For immunization, the desired protein should be available in adequate quantity (a few milligrams). However, in case of a complex multi-molecular antigen, it is quite stringent to purify it in adequate quantity. Thus, depending on its screening and selection abilities, MAbs can purify a target antigen from an antigen mixture. Mice must be immunized with antigen 6–10 weeks before fusion to allow them to develop a robust immune response before generating hybridomas. The injection schedule and the actual timing may vary depending on the antigen used for the immunization as well as other factors. It is desirable to immunize mice with a pure antigen, as this simplifies the screening of hybridomas. However, complex antigenic mixtures can be used.1–4
Collection of pre-immune serum is required prior to immunization to use as a baseline control for antibody screening. The mouse is bled by cutting approximately 1–2 mm off the tip of the tail thereby collecting 100–200 μL of blood in a capillary tube from where the serum is collected and can be cryo-preserved. A typical immunization schedule includes intraperitonial injections of 2–4 adult mice (eg, BALB/c mice) with 20–100 μg of purified antigen in a total volume of 200 μL (ie, 200 μL of a 1:1 emulsion of antigen in saline: adjuvant). A stable emulsion is critical for generating a strong immune response. The injection is repeated 14–30 days later and booster doses are administered for 2–3 times until a good titer of antibody is obtained. Next, 10–14 days after the last injection, 100–200 μL of blood is collected from the tip of the tail or from the eye and serum is separated. The antibody levels in serum can be detected by applying different immune-techniques such as ELISA, immunofluorescence, flow cytometry, and immuno blotting, and the antibody titer of the post-immune serum is compared with the pre-immune serum from the same animal. The mouse with the highest antibody titer swas selected for fusion. Between one and four days prior to fusion, the selected mouse is boosted intravenously via the tail vein. Following this step, spleen cells are prepared for fusion (Fig. 1).1–4
Myeloma Cell Line culture
In myeloma culture, hybridomas should grow continuously and selectively by suppressing the growth of the parent myeloma. It is desirable to obtain a parental myeloma cell that has been proven to yield stable hybridomas. The selected myeloma cell lines should have lost their capability to produce nucleotides using the salvage pathway. Myeloma cells are cultured in presence of 8-azaguanine so that they are unable to synthesize the HGPRT enzyme necessary for the salvage pathway of nucleic acids. Parental myeloma cells are cultured for at least one week prior to fusion to ensure that the cells are well-adapted to HGPRT-negative conditions. Cells are seeded at a density of approximately 5 × 104 cells/mL and passaged every 2 days; those growing in the early-mid log phase prior to fusion are selected for fusion (Fig. 1).1–4
After fusion, by using the drug aminopterin, de novo synthesis pathways can be blocked and thus the myeloma cells (where salvage pathway was previously blocked) cannot produce RNA or DNA and die. On the other hand, hybridomas have a functional salvage pathway (derived from the spleen cells of mouse) and can grow when they are cultured in medium containing the substrates for the pathway, ie, thymidine and hypoxanthine. This selective culture medium is HAT medium containing hypoxanthine, aminopterin, and thymidine respectively.1–4
Fusion
The parental myeloma cells used to make the hybridoma must match the strain of mouse being immunized (eg, for BALB/c mice the myeloma cells must be of BALB/c origin) and must not secrete any of their own immunoglobulin chains. The parental myeloma cells should be mycoplasma-free, fuse well, and allow the formation of stable hybridomas that continually secrete specific MAbs. SP2/0 and X63Ag8. 653 are widely used parental myeloma cells that meet all of these criteria. There are various agents that induce the somatic cell to fuse. There are some physical agents, such as electro-fusion and chemical agents, including polyethylene glycol (PEG) and calcium ions, among others. Large numbers of cells can be fused in the presence of PEG within a short time. During electro-fusion, a continuous electric potential is maintained in the fusion medium. Current is applied in short pulses at high voltage with short duration or in low voltage with long duration. The factors that are controlled during electro-fusion were specific resistance, osmotic strength, field strength, and ionic composition of the fusion medium. The cells should be given proteolytic pretreatment.1–4
An immunized mouse, 48–72 hours after tail vein injection, is euthanized and the spleen can be removed and disaggregated into a single cell suspension under sterile conditions. At the same time, the myeloma cells are harvested and added to fusion medium and mixed with spleen cells together with PEG solution to yield single hybridoma colonies. The fused cell mixture is plated in culture plates containing a feeder layer prepared from control un-immunized mice (Fig. 1).1–4
Growth and Selection of MAbs
Within 7–14 days after fusion, the growth of hybridomas occurs gradually together with the addition of interleukin 6 (IL-6), the hybridoma growth factor. Unfused myeloma cells, after 7 days of fusion, die in presence of aminopterin. Aminopterin accumulates gradually in the culture medium, while hypoxanthine and thymidine are continuously depleted, so it is essential to replace the HAT medium with hypoxanthine and thymidine (HT) medium. The qualitative and quantitative levels of the secreted antibody in the supernatant are screened by flow cytometry or ELISA. Selected cultures are cloned and re-cloned in order to achieve a pure clone population. Pure clones are grown in a larger culture flask to obtain a large amount of antibody and cells are used for future research and evaluation (Fig. 1).1–4
Long-Term Maintenance and Cryo Preservation of MAbs
The selected hybridomas are cryo-preserved in liquid nitrogen in ampules which can be thawed for use in future studies. The cryo-preserved hybridomas are thawed and re-cultured only when fresh antibody is needed. For long-term maintenance, it is always desirable to evaluate the quality of the produced antibody regularly because there may be some deterioration (Fig. 1).1–4
Antibody Production after fusion
If a large amount of antibody is required, it can be produced by growing the hybridomas in mice as an ascitic tumor, which yields ascitic fluid with antibody concentrations of nearly 1–5 mg/mL. A number of new methods are now available for purifying antibody with high purity and good yield. For some research purposes, only the supernatant of the culture medium is collected for use and induction as ascitic fluid is unnecessary (Fig. 1).1–4
Applications of Monoclonal Antibodies
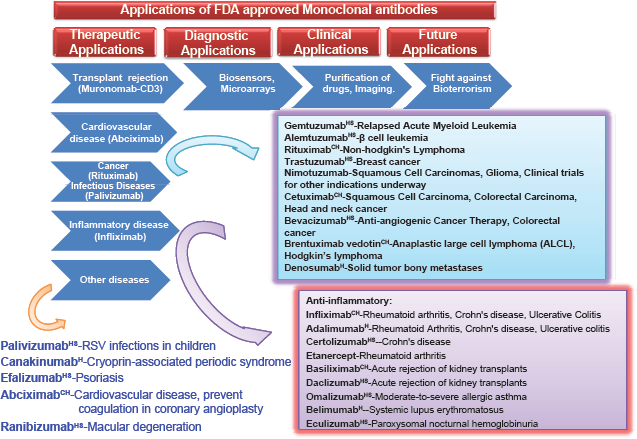
MAbs have proved to be extremely valuable for basic immunological and molecular research because of their high specificity. They are used in human therapy, commercial protein purification, suppressing immune response, diagnosis of diseases, cancer therapy, diagnosis of allergy, hormone test, purification of complex mixtures, structure of cell membrane, identification of specialized cells, preparation of vaccines, and increasing the effectiveness of medical substances (Fig. 2).1–3
Diagnostic Tools in Research and laboratory
To detect the presence of this substance/antigen, MAbs can be used. Different technologies in which MAbs are used include Western blot, immunodot blot, ELISA, radioimmuno assay (RIA), flow cytometry, immunohistochemistry, fluorescence microscopy, electron microscopy, confocal microscopy, as well as other biotechnological applications.
Gene cloning
One of the difficulties of gene cloning is identifying the cells that contain the desired gene. If an MAb that recognizes that the gene product is available, it can be used as a probe for detecting those cells that make the product and therapy to detect the gene.2,5
To Identify Cell types
MAbs contribute to the identification of many different types of cells that participate in the immune response and to unravel interactions occurring during this process. For example, in the lymphocytes with B, T helper (TH) cells and suppressor T, the use of MAbs has established that the various types of T-cells carry cell surface antigens on their surfaces that allow one type to be distinguished from another. The MAbs were also helpful in defining changes in T and B-cells during development.2,3,5
Protein purification
MAb affinity columns are readily prepared by coupling MAbs to a cyanogen bromide-activated chromatography matrix, eg, Sepharose. Since the MAbs have unique specificity for the desired protein, the level of contamination by unwanted protein species usually is very low. Since the MAb-antigen complex has a single binding affinity it is possible to elute the required protein in a single, sharp peak. The concentration of the relative protein relative to total protein in a mixture can ever be very low. This method also has limitations. Achieving 100% pure protein is difficult because there is always a tendency for small amounts of immunoglobulin to leak off the immune affinity column. Additionally, MAb do not distinguish between intact protein molecules and fragments containing the antigenic site (Fig. 2).1–3
As Therapeutic Tool in Clinical medicine
Chimeric and Humanized antibodies
Initially, murine antibodies (suffix-omab) were produced by hybridoma technology. However, the wide dissimilarity between human and murine immune systems ends in clinical failure of these antibodies, except in some specific circumstances. Murine antibodies have reduced stimulation of cytotoxicity and repeated administration of these antibodies lead to mild allergic reactions as well as anaphylactic shock, producing human anti-mouse antibodies (HAMA). These therapeutic HAMA antibodies are quickly eliminated from the host body (Fig. 3).6–8
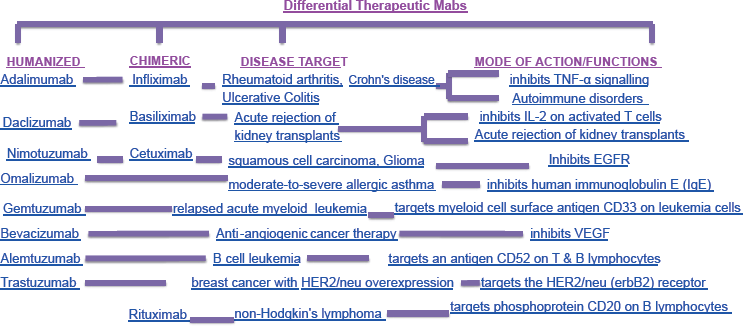
They also form immune complexes that can damage to the kidneys. To reduce the immunogenicity of murine antibodies, antibodies with increased efficiency were engineered for which the murine immunogenic content was removed. Thus, chimeric and humanized antibodies were developed. A chimeric antibody is a special type of therapeutic antibody made by combining genetic ingredients from a non-human animal (such as a mouse) and from a human. To reduce the risk of a reaction to foreign antibodies, chimeric antibodies are composed of nearly 65% human genetic material. Chimeric antibodies with their reduced immunogenicity show an increased serum half-life. In human antibodies, the murine hypervariable regions are grafted onto amino acids to produce humanized antibodies. This antibody molecule is nearly 95% human origin. Sometimes, humanized antibodies are much weaker than the parent murine monoclonal antibody in binding with antigen.6–8 Increased antibody-antigen binding affinity can be achieved with the use of techniques such chain-shuffling randomization to introduce some mutations into the complementarity determining regions (CDR). Several drugs (infliximab, rituximab abciximab, etc) based on chimeric antibodies have been approved by the Food and Drug Administration (FDA) for human use and research in this field is ever-expanding. Chimeric and humanized antibodies (such as daclizumab, omalizumab, alemtuzumab, etc) are gaining pace in for treating inflammatory diseases and cancer, among others. Several humanized and chimeric antibody products are currently available on the market for varied clinical diseases and some MAbs are going through clinical trials. Drugs made from chimeric antibodies end with the suffix–ximab and humanized antibodies are recognized by the suffix–zumab. The next stage of development in antibody production includes fully human monoclonal antibodies produced by transgenic mice or by phage display libraries. This is discussed in the antibody engineering section of this review. However, humanized MAb cannot be used effectively when the immunogen is either toxic or when there is a high degree of homology of the targeted antigen with its murine ortholog. This represents a real limitation of humanized Abs. It is some time squite convenient to develop a murine model for preclinical trial and characterization for therapeutic target, but it is not always practically applicable (Fig. 3).6–8
Diagnostic applications
The diagnostic applications of monoclonal antibodies are by far the most advanced, especially for tests that are performed on body fluids such as blood and urine sample. MAb is used to detect pregnancy as early as a week or two after conception by reacting with human chorionic gonadotrophin, a hormone secreted by the placenta and found in the urine of pregnant women. The diagnosis of veneral diseases is also being improved by the availability of MAbs. Today, MAbs are available that can identify gonorrhea (caused by Neisseria gonorrhoeae) and Chlamydia infections (caused by Chlamydia trachomatis) in 15–20 minutes, unlike the 3–7 days required by other methods. MAbs are also available to distinguish between the closely related herpes virus 1 and herpes virus 2 (Fig. 2).1–3
Identification of Cell Surface markers
Cluster differentiation (CD) and human leukocyte antigen (HLA) are expressed as cell surface molecules/antigens on various immune cells. Through flow cytometry, the CD markers or HLA molecules are identified using MAbs directed against a specific cell surface antigen. In flow cytometry, if the numbers of CD markers are under-represented, immunodeficiency disease is suspected, while if the numbers of CD markers are over-produced, cancer is indicated. Thus, calculating the expression of CD markers can be used for disease diagnosis. The MAbs have also helped to define the functions of immune cells. MAbs directed to CD4 markers on TH cells indicate the functions of T-cells. If the number of CD4 is decreased, a patient may have acquired immunodeficiency syndrome (AIDS), which may also indicate the stage of the disease. Thus, MAbs may can be used for understanding, diagnosing, and managing immune system-related diseases (Fig. 2). 3
in Detection and Localization of Intracellular antigen
Flow cytometric analysis with the aid of MAbs is primarily restricted to cell surface molecules. Thus, intracellular antigens (cytoplasmic or nuclear enzymes, cytokines, oncoproteins, immunoglobulins) were not analyzed using flow cytometry. The cytoplasmic localization of some membrane-associated molecules such as CD3 and CD22 were also not examined through cytometric studies. To localize and analyze intracellular antigens, the cells are first fixed and then perme-abilized (sometimes with detergents). Next, suitable antibody conjugates are used to localize and stain the intracellular antigen. There are some optimal processes that should be empirically determined for each intracellular antigen. In vitro cell stimulation is conducted for cytokine detection by flow cytometry as cytokine levels are mostly very low in resting cells. This stimulation of cells with proper reagent depends on the type of cell and the actual experimental conditions. 9
Cancer Diagnosis and therapy
MAbs can be used in applications against cancer cell-specific antigens that will induce an immunological response against the target cancer cell. The availability of MAbs that recognize immune cell antigens has resulted in improved diagnosis of particular types of leukemia and lymphoma. MAbs are also being applied in the diagnosis of solid tumors, particularly the carcinomas of the lung, breast, colon, and rectum. MAbs are also used to examine blood, sputum, or biopsy samples for cancer cells or for materials that have been released by cancer cells. Today, special MAbs are available for colorectal cancer, ovarian cancer, and lung cancers. MAb-mediated immunotherapy recruits cells that have cytotoxicity such as monocytes and macrophages through antibody-dependent cell cytotoxicity (ADCC). In cancer immunotherapy, MAbs binds complement proteins, which leads to direct cell toxicity that is complement dependent cytotoxicity (CDC).10–11
MAbs may also block growth factors released by tumor cells by blocking growth factor receptors, thus effectively arresting the proliferation of tumor cells. Rituximab (IDEC-C2B8), an IgG MAb is a chimeric antibody directed against the CD20 molecule and effective towards B-cell malignancies as CD20 antigen, which is present in significant numbers on malignant B-cells. Ibritumomab, an MAb against the CD20 antigen on B cells (and lymphomas), is conjugated to either the radioactive isotope indium-111 (111In) or yttrium-90 (90Y) for treatment of lymphoma patients, for who the 111In version is first given followed by 90Y version. Ibritumomab is supplemented with rituximab. Tositumomab, an MAb against CD20, is used for treating the lymphoma. 131I-Tositumomab is a single-treatment, dependable drub for B-cell lymphoma.10–13
These MAbs can also be modified for delivery of a radioisotope (radio-immunotherapy, RIT), toxin, cytokine, or other active conjugate. Bi-specific antibodies MAbs can be structured so that its Fab portion can bind to both the target antigen and to the effector cell. The FDA-regulated MAbs for cancers include bevacizumab, cetuximab, panitumumab, trastuzumab, and many others. MAbs play very crucial roles in the diagnosis of diseases such as breast cancer (block shuman cell-surface epidermal growth factor receptor-2 (HER-2) using trastuzumab. HER-2 is overexpressed in nearly 20% of breast cancers. Cetuximab, a drug used in some breast cancers and lymphomas blocks HER-1 (receptor for epidermal growth factor) on some tumor cells. MAbs may be used not only for detecting cancer cells but to destroy them, but also clinical trials have shown that MAbs have induced at least partial remission. Recent studies have shown that conjugates, drugs, and toxins with MAbs can kill leukemic cells (Fig. 2).1–3 Therapeutic anti-cancer MAbs against leukemia are gemtuzumab and alemtuzumab, rituximab for non-Hodgkin lymphoma, trastuzumab for breast cancer, and nimotuzumab and cituximab for carcinomas. Alemtuzumab confers complete remission of chronic lymphocytic leukemia by binding to a molecule (CD52) on lymphocytes. It also prevents rejection of kidney transplants.10–14
Conjugated/loaded/labeled MAbs
Conjugated MAbs are coupled with drugs and toxins and radioactive atoms are used as delivery vehicles to take these substances through the body. MAbs act as a homing device, circulating in the body until it finds a cancer cells with a matching antigen. It delivers the toxic substance to site of body where it is needed the most. This minimizes damage to other parts of the body. During chemotherapy, the delivered drugs caused damage to tumor and normal tissues.
During chemotherapy, MAbs are conjugated with chemotherapeutic drugs and are known as chemolabeled antibodies. Currently, FDA approved chemolabeled antibodies for treating cancer are brentuximab vedotin and adotrastuzumab emtansine. Brentuximab vedotin, attached to a chemo drug (MMAE) targets the CD30 antigen (present on T and B-cells) in treatment of Hodgkin lymphoma and non-responding anaplastic large cell lymphoma. Adotrastuzumab emtansine, attached to DM1 chemo drug, targets the HER2 protein antigen used for curing advanced breast cancer patients.12–15
When conjugated with toxins (poisonous substance derived from plants or bacteria), the antibodies are known as immune-toxins (eg, diptherial toxins). Denileukin diftitox, a drug used to treat some cancers (cutaneous T-cell lymphoma and many others) consists of IL-2 protein attached to a toxin (derived from the germ causing diphtheria). IL-2 normally attaches to cells that express the CD25 antigen and thus helps in delivering the toxin to these cells.12–15
Autoimmune diseases
MAbs used for autoimmune diseases include infliximab and adalimumab, which are effective in rheumatoid arthritis, Crohn's disease, and ulcerative Colitis due to their ability to bind to and inhibit tumor necrosis factor (TNF), TNF-α. Basiliximab and daclizumab inhibit IL-2 on activated T-cells and thereby help prevent acute rejection of kidney transplants. Daclizumab is also a promising drug against T-cell lymphoma. Omalizumab inhibits human IgE and is useful in moderate to severe allergic asthma (Fig. 2).1,12
The first therapeutic MAb (murine IgG 2a CD3-specific) (FDA-approved) was a transplant rejection drug, OKT3 (or muromonab, orthoclone), in 1986 is presently used in steroid-resistant patients suffering from rejection after solid organ transplant. To prevent rejection of foreign tissues, kidney transplant patients are routinely given drugs to suppress the activities of their immune systems. OKT-3 attacks the T-cells (OKT-3 antigen present in CD3) that cause the rejection.12–15
Several immune diseases are caused by an apparent attack of the immune system on the tissues of the body. Investigators are attempting to determine whether MAbs that are directed against immune cell components involved in triggering the abnormal immune responses could be used to treat such autoimmune conditions.1,6 To suppress the immune system, muromonab-CD3 (OKT3), infliximab, adalimumab, omalizumab and daclizumab are the most widely used drugs. A human MAb against Escherichia coli endotoxin has been produced which protects mice from bacteremia. It is also being tested in humans. An anti–T-cell MAb is available that has been used to remove T-cells from the donor marrow prior to transplantation, leading to reduction in graft-versus-host disease.12,13
Dental caries
In dental caries, the colonization of endogenous bacteria such as Streptococcus sp and lactobacillus sp forms the main antigen. The mucosal defense mechanism in dental caries is mediated mostly by secretory IgA (sIgA) antibodies of the saliva. Mucosal vaccination (or immunization) with purified antigen of S. mutans at induction sites helps in the migration of antigen-specific IgA to salivary glands. These IgA are produced by specific B-cells, followed by differentiation and maturation of these B-cells to secrete IgA in the saliva. Research showed that MAbs or vaccine can prevent S. mutans colonization effectively. New peptide subunits or epitopes of S. mutans have been identified, so more clinical data regarding caries experience are required before they can be used effectively against dental caries. To avoid potential disadvantages, mostly including unwanted antibody response with allergic responses, some non-human source of MAbs has been developed. 16
Other diseases
Vitaxin, a Phase II clinical trial drug shows good results in shrinking solid tumors. It has no harmful side effects. It binds to a vascular integrin (alpha-v/beta-3) present in blood vessels supplying tumors, but not on those present in normal tissues. Bevacizumab, an FDA-approved drug used in treating colorectal cancers, binds to vascular endothelial growth factor (VEGF) and thus prevents it from binding to its receptor. Vitaxin and bevacizumab act as angiogenesis inhibitors. Abciximab is used in patients who have undergone angioplasty to prevent reclogging of the coronary arteries. It inhibits the clumping of platelets by binding to its receptors.12–15
Antibody Engineering
Humanization of Murine antibodies
Anti-CD3 murine MAb (OKT-3) was approved as the first therapeutic antibody in 1986. However, OKT-3 failed as a good treatment for transplantation rejection primarily because OKT-3 as it induces severe human anti-murine antibody (HAMA) response in patients. To reduce the immunogenicity of murine antibodies in human, through genetic engineering, chimeric antibodies with human constant region and mouse variable region were constructed.1,3,9
Additionally, to reduce the effect of murine antibodies, humanized antibodies were constructed by protein engineering. In such humanized antibodies, only the complementarity determining region (CDR) loops responsible for antigen binding are grafted (by CDR-grafting) into the human variable domain framework. Simple CDR-grafting often results in significant loss of antigen binding affinity. Thus, to restore the affinity of parental murine MAb, key framework residues that support the conformation of CDR loops in the murine antibody are also grafted onto the human template. CDR-grafting has been designed with the help of computer-guided modeling of the variable domain of antibody.7,17
Generation of Fully Human Monoclonal antibodies
Apart from the routine production of murine MAbs, the production of human MAbs by conventional hybridoma technology is quite difficult because immortalized cell lines and human hybridomas do not stably produce high levels of antibody. The in vivo immunization of humans is not feasible for many antigens. However, the generation of human MAbs is made possible through the development of methods for the expression of antibody fragment (Fab or single cell variable fragment, ScFv in bacteria), the display of antibody fragments on filamentous bacteriophage and some powerful techniques for screening of antibody libraries. The phage display technique is the most used and well-established technology for the development of new human antibodies. Additionally transgenic mice containing human immunoglobulin germ line locus may be used as an alternative strategy for producing human MAbs. Immunization of transgenic mice results in a human antibody response and thus by traditional hybridoma technology, hybridomas that produce human antibodies can be generated. Humira is the first fully human MAb drug used for treatment of rheumatoid arthritis and was launched in 2003 (Fig. 3).1,2,6,7,17,18
Other Technologies involved
A method for generating a hybridoma cell without using animals has not been identified. Recent advancement of in vitro techniques allows the production of antigen-binding antibody fragments, immune library, naive library, synthetic library, antibody library screening, cell display, and ribosome display; however, these techniques are still experimental and have an uncertain result.7,17 Recently, scientists have used rabbit β-cells. Rabbits are injected with a purified antigen coupled with an adjuvant in 2–3 doses and the animal is killed and spleen cells are collected. These techniques are formulated for commercialization of monoclonal antibodies and led to a need to produce these reagents in bulk.1,2,7
Pharmaceutical Applications of MAbs
Therecent application of MAbs as a therapeutic agent has widened successful therapies for some untraced autoimmune diseases or cancers; however, proper clinical trials and FDA approval for future use must be surveyed at each and every step for its refined use. In arthritis and B-cell malignancies, antibodies were produced against the CD20 antigen, whereas in autoimmune disease and breast cancer, the well-known antibodies were generated against the inflammatory cytokine TNF as well as against HER-2 (Fig. 2). MAbs are used to produce four major classes of drugs. These classes include the first group that stimulates the body's own immune system (rituximab, infliximab etc) and the second class includes MAb drugs conjugated with a disruptive compound such as radioisotope (radio-immunotherapy, RIT). The third class includes MAb drugs conjugated with drug-activating enzyme by antibody-directed enzyme prodrug therapy (ADEPT) and the fourth classes of MAbs are conjugated to liposomes (immuno-liposomes) or to a nanotechnology drug delivery system.1–3
Most current research of RIT involves their application to lymphomas, as these are highly radio-sensitive malignancies. To limit the exposure of radiation, murine antibodies were specially choosen due to their high immunogenicity, which promotes rapid clearance from the body. For eg, to situ momab is used for non-Hodgkin lymphoma.3,12,14,19
In antibody-directed enzyme prodrug therapy, MAbs are linked to this drug-activating enzyme for its application in cancer. Subsequent systemic administration of a non-toxic agent leads to its conversion to a toxic drug, resulting in cytotoxic effects which can be delivered at malignant cells. The clinical trial of ADEPT treatments is ongoing, though it is quite promising, and can be taken in concern in future oncological treatment.2,12,10,14,19,20
Immuno-liposomes are antibody-conjugated liposomes, which carry drugs or therapeutic nucleotides and when conjugated with MAbs, may be directed against malignant cells. Although this technique is still in its formative period, significant advances have been made. Thus, immune-liposomes have been successfully used in vivo to tumors to achieve targeted delivery of tumor-suppressing genes, using an antibody fragment against the human transferrin receptor. The delivery of tissue-specific gene using immune-liposomes has also been achieved in brain and breast cancer tissue.14,19,24
Side-Effects and Limitations of MAbs
MAbs given intravenously have usually mild side effects as compared with chemotherapy. A mild allergic reaction (rash) may be occurs with first administration of the drug. Common side effects include fever, headache, weakness, chills, nausea with vomiting and diarrhea, and low blood pressure. Other side effects of MAbs are related to the targeted antigens. Bevacizumab (used against tumor blood vessel growth) can have side effects such as kidney damage, high blood pressure, bleeding with poor wound healing, and blood clots. Inhalational anthrax (potent biological terrorism) is caused by breathing the bacterial spores of Bacillus anthracis. The FDA-approved drug raxibacumab (MAb) injection is used to treat infectious inhalational anthrax when alternative therapies have failed. Common side effects include rash with itching, extreme pain, and drowsiness.9,22,23
More effective MAb drugs resulting from advancements in MAb engineering along with the development of cell biomarkers for characterizing the patient subpopulations may lead to more cost-effective use of treatment responding drugs. MAb drugs have always been costly, as only a few of nearly 22 FDA regulated drugs are available in the market. A large number of new MAb drugs are under development. The sale of first-generation safe and cost-effective MAbs is quite good in absence of generic competitors. With the high demand of specialty pharmaceuticals, some long-term measures have been taken to prominently transform the way MAbs are developed, commercialized, and marketed. MAb therapies are a financial burden on patients, and with some health plans and step-wise therapies the problem can be resolved.9,22,23
Sometimes a new, improved, costly version of an existing MAb competes with the established therapeutic product. Bevacizumab, the parent drug of ranibizumab, after usage by ophthalmologists was found to be more widely available and cheaper than ranibizumab. Additionally, it offers similar efficacy to ranibizumab. In all of these cases, comparative clinical data obtained by clinical trials is required. For a patient, the benefit of the drug does not always justify the cost, but clinical effectiveness of all these drugs should be trialed and demonstrated.22–24
MAbs is a proven therapeutic agent generating revenues of several billion dollars for the pharmaceutical industry. The typical doses of MAb drugs needed for treatment are significantly higher than those required for other drugs (or products). Thus, large-scale production that is cost-effective in manufacturing processes are required. However, the huge demand to increase production of these drugs and the drive to lower the cost of these expensive medicines is a continuous challenge to the present industry. This will further improve the efficiency of manufacturing processes. These challenges are overcome by streamlining downstream processes to increase product quantities, to implement proper quality with high-concentration product formulations with sufficient stability, dose-effective products, to reduce the cost, to develop methodologies for timeline MAb production, and to develop alternative delivery systems. The extent of glycosylation of Abs along with both cell line engineering and cell culture technologies can increase its effector function and product efficacy.22–24
Discussion
MAbs offer advantages over polyclonal antibodies due to their specificity and homogeneity between animals and breeds. Unexpected cross-reactions may occur in MAbs, whereas partial cross-reactions with common determinants take place in polyclonal antibodies. MAbs are applicable as diagnostic, imaging, and therapeutic reagents in clinical medicine and as in vitro diagnostic reagents (Fig. 2). Radio-labeled MAbs can also be used in vivo for detecting or locating disease status. Immuno-toxins composed of tumor-specific MAbs coupled to lethal toxin are being employed in tumor targeting. 19 MAbs drive the development of multibillion dollar biotechnology industry contributing to ∼32% of all revenue in the biotechnology market. Many of the leading pharmaceutical companies have entered the MAbs sector, attracted by faster and less costly development, higher success rates, premium pricing, and a potentially reduced threat from generics. The outlook for MAbs therapeutics is healthy. The ongoing success of existing products, combined with a bulging pipeline of new products awaiting approval and limited generic erosion, point towards robust growth in this segment. However, a number of technical difficulties are associated with the production of human MAbs, including lack of human myeloma cells to exhibit immortal growth and susceptibility to HAT selection, non-secretion of antibody, support of antibody production in the hybridoma, immortality of human B-cells, difficulties in of readily obtaining antigen-activated B-cells, and in vitro culture.1,2,19
In the disease myasthenia gravis, muscle weakness is a common manifestation associated with the presence of auto antibodies to the acetylcholine receptor in humans. It can be induced in rats by immunizing the rats with purified acetylcholine receptor, and then MAbs can be obtained by hybridizing spleen cells from such rats. 3 Experimental myasthenia gravis has been induced with monoclonal antibodies to acetylcholine receptors.
The importance of FDA-standardized, reproducible antibodies, with unlimited supply required for everything from tissue typing to hormone assays is a daily need for those working on applied biology. New antibodies will become available with varied diagnostic clinical and remedial values, to distinguish between different immune cell types in the course of an disease (for example, intumors); future prospects lie in locating and treatment of tumors by injecting vehicle antibody coupled with radioactive molecules or toxins.3,12,25,26
In the rejection of kidney grafts, to destroy the involved T-cells, the lymphoid system may be manipulated by injecting antibodies. In with autoimmune disease or tumors, hybridmyelomas derived from lymphocytes of patients can be used to make auto-antibodies and anti-tumor antibodies, if any, to study the disease mechanism. Auto-antibodies in mice have already been immortalized by hybridization. 3
Recently, antibodies have been used to distinguish different strains of virus for some time, and MAbs have been used for characterizing individual antigenic sites on individual components of the viruses. Additionally, antigenic variants of influenza virus have been isolated artificially by inactivating a batch of virus with MAbs and the residual variant virus ‘clones’ were grown separately. 3
However, their role in disease prediction and detection offers promise. MAb technology is a significant development where it is used as a specific serologic reagent towards antigens in unlimited amounts. These MAbs provides a highly specific and reproducible immunological assays for rapid and accurate diagnosis of an extensive list of diseases, including infectious diseases. The advantages (and disadvantages) of the use of MAbs in a number of immunoassays, such as enzyme-linked immunosorbent assays, radio-immunoassays, particle agglutination, immune-fluorescent-antibody assays (in flow cytometry, confocal, and fluorescence microscopy), and immune-histology must be evaluated. In the diagnosis of infectious disease-causing agents, a comparison of the use of MAbs and nucleic acid probe technology with respect to their perspective of applications must be reviewed. The future scope of this review remains in exploiting the potential of MAbs to replace significantly the methods currently applied in clinical laboratories.24–26
Footnotes
Author Contributions
The review was conceived, designed, data analysed and written by both authors Dr. S. Ghosh and Dr. W. Ansar and both authors have equal contribution in the review.
Funding
The study was conducted in the existing facilities of School of Biological Sciences, NISER, Bhubaneswar, DAE, Govt. of India and Post Graduate Dept. Ashutosh College, Kolkata. The authors express their gratitude to these two institutes.
Competing Interests
The authors declare no conflict of interest.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.