
Abstract
Mantle cell lymphoma is an aggressive B-cell neoplasm with special clinical and pathological characteristics. As the morphological variants of mantle cell lymphoma are currently acknowledged, a practical challenge for achieving the correct diagnosis is encountered especially when the cytogenetic/molecular data are not widely available. Here we describe a case of mantle cell lymphoma (MCL) presented with a blend of atypical clinical, morphological, and immunophenotypic features that led to an incorrect diagnosis of B-cell prolymphocytic leukemia (B-PLL).
Introduction
Mantle cell lymphoma (MCL) is an aggressive B-cell neoplasm comprising 3%–10% of non-Hodgkin's lymphomas frequently affecting middle-aged to older males with a male:female ratio of 2:1 or greater. The current 2008 World Health Organization (WHO) classification defines MCL as a distinct entity characterized by a unique immunophenotype (CD5+, CD19+, CD20+, FMC-7+, bcl-2+, cyclin D1+) and molecular translocation between the cyclin D1 gene on chromosome 11 and the immunoglobulin heavy-chain gene on chromosome 14, t(11;14) (q13;q32) with overexpression of cyclin D1.
Morphologically, it is usually composed of monotonous small- to medium-sized lymphoid cells, inconspicuous nucleoli, and slight to marked irregular nuclear contours. It is thought to arise from peripheral B-cells of the inner mantle zone of secondary follicles, mostly of naïve pre-GC type. 1 Clinically, most patients present with stage 3 or 4 disease with lymphadenopathy, hepatosplenomegaly, and bone marrow (BM) involvement. Other extranodal sites are frequently involved including the gastrointestinal tract and Waldeyer ring.1,2 Because of its aggressive clinical course, the distinction of this subtype of non-Hodgkin's lymphoma from other mature B-cell lymphoid proliferations is crucial. In clinical practice, MCL is typically diagnosed based on a combination of morphology, immunophenotype (CD20/CD5 and cyclin D1 expression), and/or the detection of t(11;14) (q13, q32) translocation. However, differentiating MCL from other B-cell lymphoid proliferations can be difficult at times as MCL may exhibit a broad morphologic spectrum that could mimic other mature B-cell neoplasms. 3 In addition, rare cases of CD5 negative,4,5 or cyclin D1/t(11;14) negative MCL has been reported. 6 Here we describe a case of MCL that presented with atypical clinical and morphological features.
Case Presentation
A 74-year-old man with a history of hypertension was admitted to the hospital for symptomatic anemia and abdominal distension in April 2010. His physical examination showed pallor and massive splenomegaly. Computerized tomography (CT) scan of the abdomen revealed massive splenomegaly measuring 23 cm and a small left lower pole renal stone. No significant lymphadenopathy was identified.
A complete blood count (CBC) revealed hemoglobin (Hb) levels of 8.9 g/dL, platelets 147 × 109/L, leukocytosis of 116 × 109/L, 88% lymphoid cells including 64% with prolymphocyte morphology. The lymphoid cells were predominantly of intermediate size with moderately dense chromatin and prominent large single nucleolus. Some of the cells had regular nuclear outlines, while others showed irregular nuclear contours (Fig. 1A). Flow cytometry (FCM) was performed on the peripheral blood showing the lymphoid cells to be positive for CD19 (bright), CD20 (bright), CD79b (bright), CD5 (bright), CD23 (moderate), and FMC7 (moderate) with Kappa light chain restriction (moderately bright). The cells were negative for CD10, CD103, and CD11c (Fig. 2). BM aspirate smears showed a predominance (76%) of atypical lymphoid cells morphologically identical to those seen in the peripheral blood (Fig. 1B). The BM core biopsy was hypercellular (approximately 70% cellularity) with interstitial infiltration by abnormal lymphoid cells and areas of diffuse infiltration. Immunohistochemical studies shows that the cells are positive for CD20, CD5, Bcl2, immunoglobulin M (IgM), IgD, and P53, with Ki-67 of approximately (40%), negative for CD10, Bcl6, DBA44, CD25, and IgG. Immunostain for cyclin D1 was essentially negative (Fig. 3). A diagnosis of chronic B-cell prolymphocytic leukemia (B-PLL) was favored taking into consideration the clinical presentation (splenomegaly), morphology, and negative cyclin D1 together with the result of the flow cytometry (positive for CD23). Fluorescence in situ hybridization (FISH) for t(11;14) was not done as it was not available at our institute at that time. Lactate dehydrogenase was slightly elevated at 538 U/L (normal 240–480); the immunoglobulin levels and electrolytes were all normal. Viral serology for hepatitis B, C, and human immunodeficiency virus were negative.
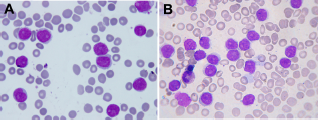
Peripheral smear (
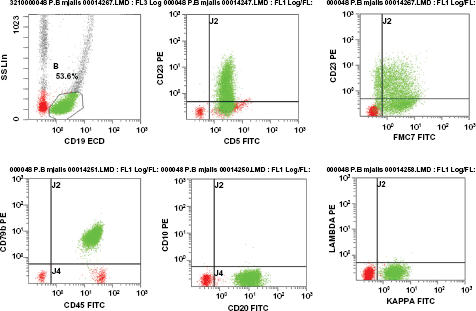
Flow cytometry; the CD19 positive cells are positive for CD5, CD20, CD79b, FMC7, CD23, and Kappa light chain restriction, and negative for CD10.
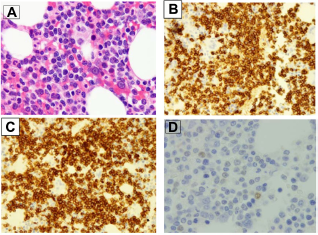
Bone marrow biopsy at presentation showing the interstitial infiltration by abnormal lymphoid cells (
The patient was started on chlorambucil 0.1 mg/kg/day for 10 days per month for 6 months. During this period he was stable, but showed no significant clinical or laboratory improvement.
On December 20, 2010 he was readmitted for fever, weight loss and night sweats of 1 month's duration. The patient was found to be pale and febrile (38.5 °C). His physical examination showed worsening of his massive splenomegaly. Septic workup and viral serology for cytomegalovirus, Epstein-Barr virus, as well as influenza A H1N1 virus and tuberculosis culture were all negative. A CT scan revealed increasing splenomegaly (26 cm), hepatomegaly (21 cm), and mild left-sided pleural effusion with left lung basal consolidation collapse. The CT scan also revealed a right paratrachial lymph node measuring 17 mm × 8 mm, and few supradiaphragmatic nodes, the largest measuring 16 mm × 7 mm. At that time, the patient's CBC showed leukocytosis of 167 × 109/L with abnormal lymphoid cells comprising 93%, with similar morphology to those seen at diagnosis. FCM on the peripheral blood showed similar findings apart from the loss of CD23 expression.
BM aspirate and biopsy was repeated with similar findings. The cells were similarly positive for CD20, CD5, Bcl2, P53, IgM, and IgD with Ki-67 of approximately 50% and negative for CD10, Bcl6, DBA44, CD25, and IgG. Cyclin D1 was also repeated and showed mostly cytoplasmic staining of the lymphoid cells with only a few of the cells showing the typical nuclear staining pattern commonly seen in the majority of cases of MCL (which typically shows nuclear staining) (Fig. 4). At this time, the suspicion of the blastic variant of MCL versus PLL was raised. All of the material was referred to a reference laboratory, which considered the cyclin D1stain as equivocal. Eventually, the cytogenetic testing by FISH for t(11;14) on a peripheral blood sample was positive in 47% of the cells, which confirmed the diagnosis of MCL (Fig. 5).
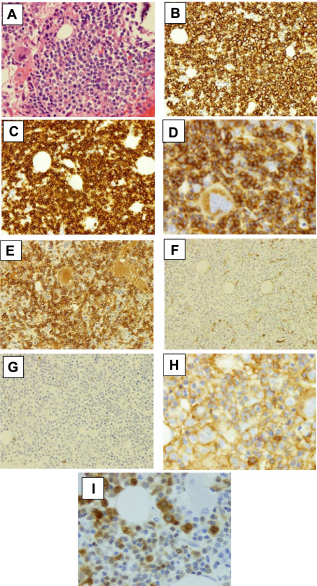
Follow-up bone marrow biopsy, showing the infiltration by abnormal lymphoid cells Haematoxylin and Eosin; ×1000 (
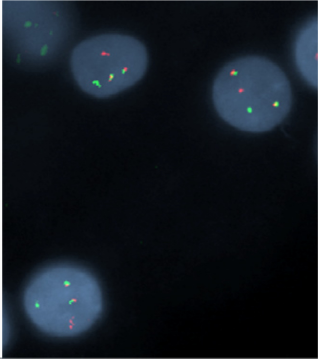
FISH for t(11;14) on a peripheral blood sample demonstrated CCND1/IGH fusion in 47% of nuclei.
The patient received supportive care and steroid, but he deteriorated rapidly and expired 1 month later with septic shock and renal failure.
Discussion
The conjunction of clinical features, cell morphology, and immunophenotyping allows for an accurate diagnosis (in most cases) of B-cell neoplasms. However, the diagnosis remains uncertain in a small percentage of cases and cytogenetic/molecular studies may help in establishing the final diagnosis. Several leukemic B-cell neoplasms may present with splenomegaly as the sole clinical finding, including PLL, 7 hairy cell leukemia, 8 splenic marginal zone lymphoma with or without villous lymphocytes, 9 and less frequently, chronic lymphocytic leukemia. 10 The differential diagnosis of these entities, presenting only with splenomegaly, is based on blood and BM lymphocyte morphology, immunophenotype, and cytogenetic/molecular studies. The subtyping is not always feasible due to lack of lymph node histologic confirmation. In such cases, a splenectomy is usually required, but this procedure carries certain difficulties in everyday practice, especially in the elderly population where these disorders are most frequently encountered.
Patients with MCL usually present with advanced-stage disease, generalized lymphadenopathy with or without organomegaly, and BM involvement. 11 A leukemic picture and extranodal disease are also frequent. 12 The mainstay of diagnosis consists of the typical morphologic, immunophenotypic, cytogenetic (FISH), and molecular characteristics of the malignant lymphocytes.
Classical MCL is generally composed of monomorphic small- to medium-sized lymphoid cells with scant cytoplasm, somewhat dispersed chromatin, inconspicuous nucleoli, and slightly to markedly irregular nuclear contours. Besides the classical type, there are some morphological variants of MCL that can be challenging with the significant morphologic overlap with other subtypes of B-cell lymphomas. In addition to the classical MCL, the 2008 WHO classification recognizes the more aggressive variants (blastoid and pleomorphic), the small cell, and the marginal zone-like variants. 1 Other rare morphological variants that overlap with follicular lymphoma, Burkitt's lymphoma 3 and prolymphocytic leukemia, 13 have also been reported. The difficulty in distinguishing B-PLL from the leukemic Blastoid variant of MCL by morphology or immunophenotye alone (without cytogenetic/molecular studies or cyclin D1 positivity) is acknowledged since the peripheral blood and marrow morphology, as well as the extended immunophenotype, is insufficient for an accurate diagnosis, especially since the differences are slight and challenging even for an expert panel. The distinction between the two entities is particularly problematic when patients present with isolated splenomegaly, as well as with no lymphadenopathy or extranodal involvement, similar to the case we are reporting.
It has been shown that a substantial proportion of cases previously classified as t(11;14) positive B-PLL should be reclassified as MCL, and this has important prognostic and treatment implications as MCL has a poor median survival and requires more aggressive treatment. 14
Ruchlemer et al 14 reclassified eight cases that were diagnosed before 1995 as B-prolymphocytic leukemia harboring t(11;14) translocation and/or cyclin D1 staining as MCL following the review of histopathological, immunophenotypic, and cytogenetic data. In these eight cases the clinical characteristics were similar to a t(11;14)-negative B-PLL control group with significant overlap in the peripheral blood and BM morphology. Although CD5 expression was more commonly encountered in the translocation positive group with stronger surface immunoglobulin expression and less frequent expression of CD23, these differences were not statistically significant. However, the spleen histology was different, and a population of atypical mantle cells was found in all of the t(11;14)-positive cases.
Cyclin D1 overexpression is believed to be essential in the pathogenesis of MCL. Immunostaining for cyclin D1 is considered a convenient initial step in the workup, as the result can be obtained quickly. The existence of cyclin D1-negative MCL has been controversial and difficult to substantiate since cyclin D1 overexpression is believed to be essential in the pathogenesis of MCL. However, it is plausible that cases of cyclin D1- negative MCLs do exist and are part of the spectrum of MCL heterogeneity.
In the last few years, the gene expression profiling of large numbers of MCL cases has allowed for the identification of a particular subset of tumors with the same clinical presentation, morphology, phenotype, and global expression profile as conventional MCL, except for the lack of cyclin D1 overexpression and the t(11;14) translocation. These tumors were termed cyclin D1-negative MCL. Kai Fu et al 6 reported six cases of cyclin D1-negative MCL. All six cases exhibited the characteristic morphologic features and the unique gene expression signature of MCL, but all were negative for cyclin D1 by immunostaining and they lacked cyclin D1 mRNA expression by both quantitative reverse transcription polymerase chain reaction and gene expression analysis. They also lacked the t(11;14) (q13; q32) by FISH analysis. Interestingly, these patients overexpressed cyclin D2 (two cases) or D3 (four cases), suggesting that the upregulation of these G1 cyclins could be an alternative mechanism in the pathogenesis of MCL. The similar secondary genetic profiles and alterations observed in cyclin D1-positive and cyclin D1- negative tumors supports the idea that they correspond to the same biologic entity and share similar genetic pathways of evolution. 15
Most reported cases of cyclin D1-negative MCL have been attributed to suboptimal immunostaining, inadequate genetic or molecular analysis, or misdiagnosis.
Immunostaining for cyclin D1 is considered as an important adjunct for the diagnosis of MCL, particularly when the stain is applied on lymph nodes and other soft tissue. Nevertheless, immunohistochemical demonstration of cyclin D1 may be unreliable, and several limitations and technical problems were reported. Insufficient epitope retrieval, too low concentrations of the primary antibody, and the use of low affinity antibodies were reported as the most frequent causes of false negative cyclin D1 reactions. 16 Difficulties with staining of decalcified BM specimens were also acknowledged and unfortunately, fixation and decalcification of the BM biopsy may jeopardize the immunohistochemical demonstration of cyclin D1. Fixation in a 1% formaldehyde solution containing 0.4% glutaraldehyde was found to completely abolish staining for cyclin D1. 17 High background staining interfering with interpretation has been reported by Vasef et al, 18 and they also showed that only 72% of involved marrow had cyclin D1 overexpression according to immunohistochemistry. An overall weaker intensity of reactivity was reported in the marrow compared to the lymph node and spleen. 19 Torlakovic et al 20 compared the performance of five anti-cyclin D1 antibodies (monoclonal SP4, P2D11F11, and DCS-6, and polyclonal CP236 and 06-137) on multi-tissue blocks, and reported that the polyclonal CP236 has the highest sensitivity (100%) followed by SP4 (95%).
In our case, cyclin D1staining was conducted on the first and the second marrow samples with the monoclonal antibody SP4 (Flex prediluted; Dako, Glostrup, Denmark), Dako Target Retrieval HIGH PH 9, and Dako Envision Fex detection reagent using Dako autostainer Link 48. The fixative used for the marrow biopsy was Bouin's, which might contribute to the false negative stain on the first sample as deleterious effects on some immunostains, including cyclin D1 was reported with Bouin's fixative (a formalin-based solution with picric acid and acetic acid). 21
Pathologists should be familiar with the typical morphology of MCL, atypical variants, and its mimics to decrease or prevent diagnostic errors. Careful morphologic examination, with knowledge of the full spectrum of morphological variants in MCL, is critical when suspecting a diagnosis of cyclin D1-negative MCL, or those with equivocal cyclin D1 immunostaining. This diagnosis may be challenging, particularly in cases with atypical features, a challenge which was encountered in our case as it combines atypical clinical, morphological, and immunophenotypic features in addition to the negative cyclin D1 at presentation.
The choice of proper fixative, high affinity antibodies are crucial for cyclin D1 staining, and it might be prudent to have two highly sensitive antibodies in the laboratory; when one gives a negative reaction, the other probably should be used for confirmation.
Our case also emphasizes the importance of incorporating clinical data, morphology, immunophenotyping, and cytogenetic/molecular studies in diagnosing and classifying lymphomas.
Footnotes
Author Contributions
Main author performed the diagnostic part and formulated the paper: FI. Wrote the clinical aspects of the paper: HEO. Interpretation of the clinical data and helped draft the discussion and the abstract: RYT. Reviewed the paper IAH. Reviewed the manuscript: MEK, AAS.
Funding
Author(s) disclose no funding sources.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosure and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest. Institutional approval protocol: 11201/11.