
Abstract
Aim
This study was designed to examine the immunogenetic basis for shared autoimmunity, resulting in autoantigen presentation that leads to the production of two or more disease-specific autoantibodies.
Methods
A bioinformatics approach based on peptide binding predictions to disease-associated HLA determinants has been developed and tested here using 11 disease associations between autoimmune systemic and mucocutaneous blistering disorders. Various HLAs associated with antigens within a given “disease model” (set of HLA class II and protein sequences known to be associated with a specific autoimmune disease) were tested and ranked against the antigenic proteins, first with proteins they are known to associate with and then with proteins known to be implicated in a second disease model. In every case binding predictions were compared for different proteins binding to the same HLA. Subsequently, disease-related autoantigens have been tested for their binding affinity against each disease-specific HLA class II protein.
Results
For a single HLA haplotype, several binders have been generated from a related autoantigen with the variable binding score. In most cases, the binding score corresponding to the interactions between the autoantigen-derived epitope and the HLA associated with one disease was similar or lower than the interactions between the epitope from proteins associated with the second disease and the same HLA. Notably, there was no compelling promiscuity in peptide binding to each of the HLA molecules, in spite of the promiscuous nature of HLA class II binding.
Conclusions
The data suggest that, in susceptible individuals, shared autoimmunity might be initiated by two types of HLA/peptide interaction; first between an autoantigen-derived epitope and its disease-associated HLA molecules, and second, between a different peptide of the same autoantigen and HLA proteins specific for the second disease.
Keywords
Introduction
Autoimmune mucocutaneous blistering diseases (AMBD) such as pemphigus vulgaris (PV), pemphigus foliaceus (PF), bullous pemphigoid (BP), ocular cicatricial pemphigoid (OCP), dermatitis herpetiformis (DH) and mucous membrane pemphigoid (MMP), are a group of rare organ-specific diseases that affect skin and multiple mucous membranes.1–5 PV is a potentially fatal disease characterized by the loss of intercellular adhesion of keratinocytes, resulting in acantholysis.6–8 In the serum of PV patients, high titers of circulating autoantibodies targeting the epidermal adhesion molecule desmoglein 3 (Dsg3)–-keratinocyte transmembrane proteins localized in the desmosome, essential for maintaining the integrity of the epidermis–-are believed to cause clinical disease by direct binding to and disruption of desmoglein proteins.1,9 The association of HLA class II antigens with susceptibility to PV has been demonstrated in numerous studies.10–14 In Ashkenazi Jews, PV appears tightly linked to the rare haplotype HLA-DR4 (DRB1*0402): DQB1*0302, while in non-Jewish patients, it is linked to the haplotype DRB1*404X: DQB1*0503. 15
MMP affects mucous membranes of the body and is characterized by the presence of autoantibodies to human β4 integrin,16,17 while BP which predominantly affects the skin and is associated with bullous pemphigoid antigen 1 (BPAg1) and 2 (BPAg2). 18 Both BP and MMP have been shown to have a strong linkage to DQB1 *0301.18,19 It has been demonstrated that the same patient may have antibodies against more than one autoantigen within the skin and mucous membrane, resulting in more than one autoimmune mucocutaneous disease. For example, patients with PF may develop BP,20,21 patients with MMP may have PV (22), and some patients are affected with both PV and OCP. 23
OCP is a systemic autoimmune pemphigoid disorder that has both ocular and non-ocular manifestations. OCP can cause bullous lesions of the skin and mucous membranes that result in scarring of the affected skin, conjunctiva (inner lining of the eye), and other mucous membranes.24,25 Different epithelial membrane zone components have been recognized by antibodies in patients with OCP, ie, BPAg1 and BPAg2, laminin 5, laminin 6, type VII collagen, β4 integrin subunit, and antigens with unknown identities (a 45-kd protein, uncein, a 168-kd epithelial protein, and a 120-kd epithelial protein).26,27 Among white patients in the United States, OCP is associated with the DQB1 *0301 allele.
DH is an autoimmune blistering disorder associated with a gluten-sensitive enteropathy. It is characterized by grouped excoriations, erythematous, urticarial plaques, and papules with vesicles. DH is caused by the deposition of IgA in the papillary dermis, which triggers an immunologic cascade, resulting in neutrophil recruitment and complement activation.28,29 DH is associated with an increased expression of HLA-A1, HLA-B8, HLA-DR3, and HLA-DQ2 haplotypes.30,31 Evidence is mounting that epidermal transglutaminase 3 (TGM3), a cytosolic enzyme involved in cell envelope formation during keratinocyte differentiation, is the autoantigen of DH. Theoretically, DH is caused by dermal deposition of circulating immune complexes containing both IgA and TGM3.32,33
In contrast to these organ-specific diseases, connective tissue disorders, or systemic diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and systemic sclerosis (SSc), involve multiple tissues and organs.34–36 Mixed connective tissue disease (MCTD) is also a systemic autoimmune syndrome that is characterized by the presence of high titers of serum antibodies against small nuclear ribonuclear proteins (U-snRNPs),37,38 in particular against U1 small nuclear RNP polypeptide (U1 snRNP). It has been suggested that MCTD represents a distinct clinical entity, based on clinical manifestations that separate MCTD from other connective tissue diseases. 39 Various associations of HLA class II antigens with MCTD have been reported, including HLA-B7 and HLA-Dw1. 40 In another study, HLA-DR4 was found to be significantly increased in MCTD, 41 whereas others reported an association between HLA-DQw3 and anti-RNP antibodies in patients with MCTD. 42 Interestingly, MCTD patients with increased IgG autoantibodies against U1-70 kD polypeptide have an increased prevalence of HLA-DR4 compared with controls. 43 Furthermore, molecular biology studies have shown that most MCTD patients carrying HLA-DR4 or HLA-DR2 alleles share a region of homology consisting of seven amino acids in the HLA-DRB1 gene. 44 This “shared epitope” of HLA-DR molecules, in different alleles and in different patients with MCTD, may be important for the modulation of the autoimmune response to the U1-70 kD antigen. 45
Based on the accumulated evidence of shared autoimmunity, it has been intriguing to investigate the relationship between the genetic and immunological mechanisms for the simultaneous production of two or more autoantibodies in patients exhibiting more than one autoimmune disease. A hypothesis, which in part may explain some of the increased susceptibility to both autoimmune blistering and systemic connective tissue diseases, has recently been proposed. 46 This hypothesis is based on the unequivocal evidence that CD4+ T cells play a critical role in autoantibody production, implying that existence of HLA class II-restricted T cell epitopes might trigger autoimmunity. Three possible scenarios for the immunopathogenic mechanisms leading to breakage of tolerance and induction of two distinct autoimmune diseases in the same individual have been discussed: 1) T cell epitopes of the two different autoantigens associate with each of the susceptible HLA molecules, resulting in dual autoimmunity; 2) a single epitope of an autoantigen binds to both HLA specificities, leading to the induction of both diseases by cross- reactivity; and 3) two distinct epitopes of the same autoantigen are able to bind two different HLA molecules that are associated with the two diseases, implying that both T cell epitopes originating from one autoantigen will activate immunopatogenic mechanisms by binding to two HLA molecules specific for two diseases. In order to test each of these hypotheses, bioinformatics-based predictions of peptide binding to disease-associated HLA determinants have been generated using multi-HLA II peptide binding analysis. This communication explores the nature of autoantigen-derived epitopes and their binding characteristics to HLA proteins in the context of shared autoimmunity.
Methods
Protein sequences
The protein sequences of autoantigens were collected from the Consensus Coding Sequence Database (CCDS), a collaborative effort between the NCBI and several other organizations which contains consistently annotated protein coding regions of human and mouse genes. 47 Only human sequences were used in this project, and alternate exons were included in the sequences. The autoantigens tested in this study are listed in Table 1, and in most cases consist of the entire protein sequence from which specific autoantigens (such as BP180 and BP230) are derived.
Autoantigens and HLA associations in autoimmune blistering and systemic diseases.

Peptide-HLA II binding predictions
The analysis of peptide binding to HLA II molecules known to be involved in autoimmune mucocutaneous blistering diseases was based on the use of the same methodology applied by the Rankpep web server. 48 Rankpep predicts peptide binding using position specific motif matrices–-also known as motif profiles–-that are derived from known peptide binders.49,98 A program was written, cross-HLA II-binding analyzer, to perform multiple rankings of predicted binding potentials and classify the binding predictions into sets: predictions for protein-HLA II sets known to be involved in disease genesis. Results were organized by disease model–-sets of HLA II and proteins known to be involved in the genesis of specific autoimmune diseases were used.
The cross-HLA II-binding analyzer performs three types of sorting based on the three scenarios proposed in this paper: 1) Single HLA II recognition, 2) Dual HLA II recognition with a single epitope, and 3) Dual HLA II recognition with dual epitopes. The analysis was designed to group the components of each disease model, the HLA IIs and proteins involved in known disease interactions–-for example, HLA-DQB1*0301 with alpha 1 type XVII collagen (BP180) and dystonin (BP230) in the BP model–-and sort results by within-model and cross-model HLA II-peptide binding pairings. Some details of the cited scenarios are as follows:
Scenario 1–-Single HLA recognition: This scenario is assumed to be the standard case, since each disease model exists independently. The cross-HLA II-binding analyzer generates a series of ranked results within the known disease models described above (sets of HLA II and proteins known to contain antigenic sequences), showing all predicted binders for each HLA II-protein pairing–-peptides scored above the binding threshold for that pairing. Because these interactions are within known disease models, they are used as reference values.
Scenario 2–-Dual HLA II recognition with a single epitope: the cross-HLA II-binding analyzer has an intersection analysis function, which is used to find promiscuous peptides in a series of two HLA II-peptide binding predictions. To identify promiscuous peptide-binding peptides, the analysis was limited to the set of the top 5% of peptides in each group of results. This analysis identifies promiscuous peptide binders across known disease models. When a common peptide was found in more than one autoantigen, it was included in an intersection analysis report, detailing the binding score of the common peptide for each matrix.
Scenario 3–-Dual HLA recognition with dual epitopes: the cross-HLA II-binding analyzer produces results for cross-model pairings of HLA IIs and proteins. To evaluate the potential for the pairings to produce immunogenic reactions, binding predictions (binding affinity, number of predicted binders) across models are compared to the top predicted binding scores for known immunogenic reactions within known disease models (Scenario 1).
Results
Based on the published observations of shared autoimmunity and on theoretical predictions exploring the probability of its occurrence, the following disease associations have been tested: PV/MMP, PF/BP, PV/OCP, PV/BP, PV/MCTD, PF/MCTD, BP/MCTD, DH/MCTD, OCP/MCTD, MMP/MCTD and BP/DH. The list of autoantigens and HLA associations in autoimmune blistering and systemic diseases is presented in Table 1. For most diseases in this study, eg, PV, MMP, DH, PF and MCTD, a single autoantigen has been reported, with the exception of BP and OCP that are characterized by the presence of the same two autoantigens, BPAg1 (BP180), derived from alpha 1 type XVII collagen and BPAg2 (also known as BP230), part of the dystonin protein. It is noteworthy that MMP, BP and OCP are all associated with the prevalent HLA-DQB1*0301 allele, while PV, DH, PF and MCTD each carry distinct HLA haplotypes.
Three hypothetical models of peptide-HLA II interactions proposed by Fridkis-Hareli 46 were tested using HLA-specific binding matrices and autoantigen sequences listed in Table 1. For each HLA specificity, the autoantigen of interest was examined for the presence of potential binding epitopes and scored based on peptide binding affinity. The results arranged by disease model are shown in Supplemental Tables 1–11. All potential epitopes (sequences predicted to bind above the binding threshold for the HLA matrix were retrieved from each analysis (raw data not shown). It is important to note that comparisons were made between HLA-peptide binding analyses against the same HLA molecule–-all binding scores and values were relative to that specific HLA class II. It should also be noted that not all the HLA specificities known to associate with a particular disease were available from the database and thus were not included in the present report.
PV/MMP
Autoantigenic proteins associated with each of these diseases were tested for binding to the HLA haplotypes as described below:
PV Model: the HLA II-peptide biding analysis for desmoglein 3 preproprotein generated 991 sequences in total (Supplemental Table 1). Four Dsg3- derived peptides were predicted to bind to HLA-DQ8 (DQA1*0301/DQB1*0302) above the binding threshold, with a predicted range of 36.91%–25.22% (from high to low optimal binding). In contrast, binding of the same autoantigen to HLA-DR4 (DRB1*0402) determinants resulted in 11 predicted binders with a range of 40.33%–25.80% of the optimal score. Similarly, five predicted binders of Dsg3 to HLA-DR4 (DRB1*0404) were identified with a range of 53.46%–32.23% of the optimal score (Supplemental Table 1A). The sequences of Dsg3-derived binding epitopes are presented in Supplemental Table 1B.
MMP Model: Table 2 shows that the analysis for HLA-DQ7 (DQB1*0301) and β4 integrin generated 1814 sequences. Fourteen peptides were predicted to bind, at 44.19%–25.90% of the optimal binding score. For peptide sequences refer to Supplemental Table 1B.
Predicted binding epitopes derived from autoantigens specific for blistering and systemic disease combinations.
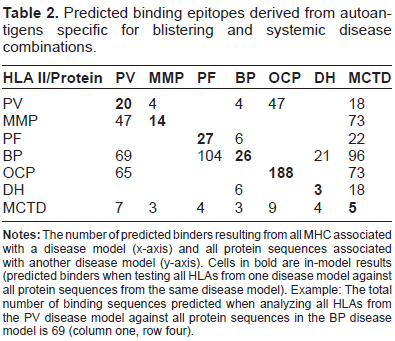
PV/MMP: The cross-model analysis matches HLA-DQ7 (DQB1*0301) (from the MMP model) with Dsg3, the antigen-producing protein from the PV model. Four sequences from Dsg3 are predicted to bind, with scores between 37.32% and 29.70% of the optimal score for HLA-DQ7 (DQB1*0301). These sequences are within the range of binders predicted for the in-model interactions, but four of the predicted in-model binders have higher scores than the best cross-model prediction (Supplemental Table 1A).
Conversely, the cross-model analysis matches β4 integrin (MMP model) with HLA-DQ8 (DQA1*0301/DQB1*0302) (14 predicted binders), HLA-DR4 (DRB1*0402) (16 predicted binders) and HLA-DR4 (DRB1*0404) (17 predicted binders) from the PV model. Interestingly, the two highest scoring peptides from the HLA-DQ8 (DQA1*0301/DQB1*0302)–-β4 integrin (DERCHLDTT and ANRCKKAPV), scored higher than all the predicted binders from the PV in-model analysis. In contrast, the binding of β4 integrin to HLA-DR4 (DRB1*0402) and HLA-DR4 (DRB1*0404) was within the binding range of Dsg3 to these molecules (Supplemental Table 1B).
PF/BP
In this case, three autoantigens, ie, Dsg1 for PF, alpha 1 type XVII collagen (BP180) and dystonin (BP230) for BP have been analyzed for their binding to several disease-associated HLA haplotypes, as shown in Supplemental Table 2. Analysis of Dsg1 binding to PF-associated molecules revealed 1041 sequences in total, whereas alpha 1 type XVII collagen (BP180) generated 1489 sequences and dystonin (BP230) generated 2641 sequences derived from their binding to BP-associated MHC molecules (Supplemental Table 2A).
PF Model: Peptide binding analysis for desmoglein 1 (Dsg1) to HLA-DR1 (DRB1*0101) showed 22 results with 53%–16% predicted optimal binding. In addition, analysis of Dsg1 and HLA-DR4 (DRB1*0404) identified five binders in the range of 48%–32% of predicted optimal values. In contrast, analysis for Dsg1 binding to HLA-DR4 (DRB1*0406) and HLA-DR1 (DRB1*0102) showed no binding epitopes above the binding threshold (Supplemental Table 2A). Peptide sequences are shown in Supplemental Table 2B.
BP Model: HLAII-peptide binding results for alpha 1 type XVII collagen peptides binding to HLADQ7 (DQB1*0301) showed 14 peptide sequences ranging between 43%–26% optimal binding (Supplemental Table 2A). Binding of dystonin to the same molecules resulted in 12 predicted binders with 31%–26% of optimal binding (Supplemental Table 2A).
PF/BP: Analysis of Dsg1 binding to BP-associated HLA-DQ7 (DQB1*0301) molecules resulted in six peptides with 39%–28% optimal binding (Supplemental Table 2A). Notably, these values were lower than the binding of Dsg1 to PF-associated MHC molecules. Predictions for alpha 1 type XVII collagen peptides binding to PF-associated HLA-DR1 (DRB1*0101) resulted in 30 binders with 40%–17% optimal binding (Supplemental Table 2A). In contrast, analysis of alpha 1 type XVII collagen/HLA-DR1 (DRB1*0102) interactions showed no detectable binding. On the other hand, the binding of alpha 1 type XVII collagen to HLA-DR4 (DRB1*0404) showed higher range of optimal binding (52%–35%) for six predicted epitopes (Supplemental Table 2A). Interestingly, predictions of alpha 1 type XVII collagen binding to HLA-DR4 (DRB1*0406) resulted in one peptide epitope with the binding score of 36.062 and optimal binding of 49.23% (Supplemental Table 2A). Peptide sequences are listed in Supplemental Table 2B.
Binding of dystonin to PF-associated HLA-DR1 (DRB1*0101), HLA-DR1 (DRB1*0102), HLA-DR4 (DRB1*0404), HLA-DR4 (DRB1*0406) was tested similarly to alpha 1 type XVII collagen. For HLA-DR1 (DRB1*0101) molecules, 56 predicted binders with 44%–16% optimal binding have been identified, and for HLA-DR4 (DRB1*0404), 11 dystonin-derived binders with 54%–32% optimal binding have been detected (Supplemental Table 2A). Of note is that these values are higher than the ones found for dystonin binding to BP-associated molecules. Interestingly, no predicted epitopes were found either for HLA-DR1 (DRB1*0102) or for HLA-DR4 (DRB1*0406) interactions with dystonin. In summary, these results suggest that here, as in the case of PV/MMP, epitopes derived from one disease-specific autoantigenic protein may bind both disease-specific HLA molecules.
PV/OCP
Analysis of Dsg-3-derived peptides to PV-associated HLA-DQ8 (DQA1*0301/DQB1*0302), HLA-DR4 (DRB1*0402) and HLA-DR4 (DRB1*0404) molecules has been described in PV section (Supplemental Table 1). Likewise, due to the fact that OCP shares the same autoantigens and HLA specificities with BP and MMP, ie, alpha 1 type XVII collagen, β4 integrin and HLA-DQ7 (DQB1*0301), the corresponding data have been provided in BP and MMP sections (Supplemental Tables 1 and 2). Analysis of β4 integrin binding to OCP-associated MHC molecules generated 1814 peptide sequences, whereas alpha 1 type XVII collagen generated 1489 sequences (Supplemental Table 3A).
PV/OCP: PV-specific desmoglein 3 preproprotein peptide binders examined for the OCP-associated HLA-DQ7 (DQB1*0301) revealed four binders with 37%–29% predicted optimal binding, as described for PV/MMP model (Supplemental Table 1A). The binding of Dsg3 to HLA-DR4 (DRB1*0401) resulted in 43 binders with 46%–11% predicted optimal values (Supplemental Table 3A). In the case of OCP-related autoantigen association with PV-specific MHC, eight binders derived from alpha 1 type XVII collagen (BP180) exhibited 36%–25% optimal predicted binding to HLA-DQ8 (DQA1*0301/DQB1*0302). The binding to HLA-DR4 (DRB1*0402) was between 38%–28% optimal values for four predicted binders, while the binding to HLA-DR4 (DRB1*0404) was even higher (52%–34% optimal binding). Another potential OCP-associated autoantigen, β4 integrin, bound to PV-specific HLA molecules with differential predicted scores as shown in Supplemental Table 3A. For HLA-DQ8 (DQA1*0301/DQB1*0302), 14 binders with 42%–25% optimal binding have been identified, whereas 16 binders were detected for HLA-DR4 (DRB1*0402) (37% optimal binding), and 17 peptides for HLA-DR4 (DRB1*0404) with 51%–34% optimal binding (Supplemental Table 3A). Interestingly, the binding of β4 integrin to PV-associated HLA-DR4 (DRB1*0404) was higher than to OCP-associated molecules (51% vs. 44%, respectively). Peptide sequences are shown in Supplemental Table 3B.
PV/BP
For this disease combination, individual parts of PV/Dsg3 and BP/alpha 1 type XVII collagen/dystonin analysis have been described in PV and BP sections, respectively (Supplemental Tables 1 and 2).
PV/BP: Analysis of the binding of Dsg3 to BP-associated HLA-DQ7 (DQB1*0301) resulted in four binders with 37%–29% optimal binding (Supplemental Table 4A). In the opposite direction, the binding of alpha 1 type XVII collagen to PV-associated HLA-DR4 (DRB1*0402), HLA-DR4 (DRB1*0404), and HLA-DQ8 (DQA1*0301/DQB1*0302) showed four binders (38%–28% optimal binding), six binders (52%–34% optimal binding) and eight binders (36%–25% optimal binding), respectively. For dystonin-derived peptides, 28 predicted binders were found with DRB1*0402 (37%–28% optimal binding), 11 peptides bound to DRB1*0404 (54%–32% optimal binding), and 12 predicted binders (47%–25% optimal binding) were detected for HLA-DQ8 molecules (Supplemental Table 4A). Of note is that the binding score for dystonin-derived peptides bound to PV-associated HLA-DQ8 molecules was higher than the one for dystonin binding to BP-associated HLA-DQ7 molecules (Supplemental Table 4A). Peptide sequences are presented in Supplemental Table 4B.
PV/MCTD
PV-related Dsg3 association with its HLA-DQ8 (DQA1*0301/DQB1*0302) receptors was already described in this report (Supplemental Tables 1, 3 and 4). Peptide binding analysis of MCTD-specific autoantigen UI-snRNP C [Homo sapiens] resulted in 429 sequences in total (Supplemental Table 5). Binding of UI-snRNP-derived peptides to HLA-DR1 (DRB1*0101) revealed three predicted epitopes with 37%–25% optimal binding, while interaction with the 9mer HLA-B07 resulted in two peptides with 63%–62% optimal binding (Supplemental Table 5A). PV-related Dsg3 association with its HLA-DQ8 (DQA1*0301/DQB1*0302) receptors was already described in this report (Supplemental Tables 1, 3 and 4).
PV/MCTD: The combination of PV and MCTD was tested using autoantigens UI-snRNP and Dsg-3, and HLA molecules associated with both diseases, ie, HLA-DR1 (DRB1*0101), HLA-DR4 (DRB1*0402), HLA-DR4 (DRB1*0404), HLA-DR4 (DRB1*0406), HLA-DQ8 (DQA1*0301/DQB1*0302) and HLA-B07 alleles. The binding of PV-specific Dsg3 to MCTD-associated HLA-DR1 (DRB1*0101) showed 18 binders with 44%–17% optimal binding, while analysis of Dsg3/9mer-HLA-B07 interactions showed no predicted binding epitopes (Supplemental Table 5A). Similarly, there was no binding of UI-snRNP to PV-associated HLA-DR4 (DRB1*0404), but three peptides (55%–26% optimal binding) were found to bind HLA-DR4 (DRB1*0402), and four peptides (40%–27% optimal binding) were detected for HLA-DQ8 (DQA1*0301/DQB1*0302) molecules (Supplemental Table 5A). Importantly, the optimal binding of UI-snRNP-derived peptides to PV-associated HLA-DR4 (DRB1*0402) was higher than its binding to MCTD-related HLA-DR1 (DRB1*0101) but lower than binding to the second susceptible MCTD allele, HLA-B07 (Supplemental Table 5). Peptide sequences are shown in Supplemental Table 5B.
PF/MCTD
Binding analysis of PF- and MCTD-related peptides to their disease-specific HLA molecules was described in the earlier sections of this report (Supplemental Tables 2 and 5). The largest number of Dsg1-derived epitopes was found to bind to HLA-DR1 (DRB1*0101) as compared to other PF-specific alleles (Supplemental Table 6). This disease combination is unusual since both diseases share the presence of HLA-DR1 (DRB1*0101), suggesting that PF- and MCTD-derived peptides might bind to the same molecule and thus trigger initiation of the second disease.
PF/MCTD: Binding analysis of Dsg1-derived peptides to MCTD-associated HLA-DR1 (DRB1*0101) molecules showed 22 epitopes (53%–16% optimal binding), while no binders were found for the 9mer-HLA-B07 (Supplemental Table 6A). Predictions for UI-snRNP-derived peptides bound to HLA-DR1 (DRB1*0101), HLA-DR1 (DRB1*0102), HLA-DR4 (DRB1*0404) and HLA-DR4 (DRB1*0406) molecules showed three (38%–25% optimal binding), zero, zero, and one binders (35% optimal binding), respectively (Supplemental Table 6A). Interestingly, the binding of Dsg1-derived peptides to HLA-DR1 (DRB1*0101) was higher than that of the UI-snRNP-derived peptides as expressed by the optimal binding values. Peptide sequences are shown in Supplemental Table 6B.
BP/MCTD
Binding of alpha 1 type XVII collagen and dystonin to BP-associated HLA-DQ7 (DQB1*0301) molecules, as well as the binding of UI-snRNP to MCTD-associated HLA-DR1 (DRB1*0101) and the 9mer-HLA-B07 has been described in the previous sections of this study and is shown in Supplemental Tables 2, 4, 5 and 6.
BP/MCTD: Binding analysis of alpha 1 type XVII collagen peptides to MCTD-specific HLA-DR1 (DRB1*0101) resulted in 30 predicted peptides (40%–17% optimal binding), while the binding to the HLA-B07 molecules showed nine binders (59%–45% optimal binding) (Table 7). Moreover, the binding of dystonin-derived peptides to HLA-DR1 (DRB1*0101) showed 56 peptides (44%–17% optimal binding), and only one peptide (53% optimal binding) for the 9mer-HLA-B07 molecules (Supplemental Table 7A). Interestingly, three UI-snRNP-derived peptides were shown to bind to BP-associated HLA-DQ7 (DQB1*0301) molecules (33%–26% optimal binding). Here, the binding of BP- and MCTD-associated autoantigens to their disease molecules was similar to the cross-disease combination. Peptide sequences are presented in Supplemental Table 7B.
DH/MCTD
Analysis of DH-specific TGM3 transglutaminase 3 peptide binders resulted in 685 peptide sequences (Supplemental Table 8). Binding predictions for TGM3-derived peptides to HLA-DQ8 (DQA1*0301/DQB1*0302) showed three binders (30%–28.5% optimal binding), whereas no binders were found for HLA-DQ2 (DQA1*0501/DQB1*0201) (Supplemental Table 8A). Studies of UI-snRNP/HLA-DR1 (DRB1*0101)/9mer-HLA-B07 have been described in the previous sections (Supplemental Tables 5–7).
DH/MCTD: Binding analysis of TGM3 to HLA-DR1 (DRB1*0101) alleles resulted in 17 binders (41%–17% optimal binding), but only one binder was found for the 9mer-HLA-B07 molecules (optimal binding 47.5%). In the opposite direction, the binding of UI-snRNP to HLA-DQ2 (DQA1*0501/DQB1*0201) showed four peptides (40%–27% optimal binding) and no binding to HLA-DQ8 (DQA1*0301/DQB1*0302) molecules (Supplemental Table 8A). Peptide sequences are shown in Supplemental Table 8B.
OCP/MCTD
For a single disease model, analysis of OCP- and MCTD-specific peptides bound to their associated HLA molecules has been shown earlier (Supplemental Tables 3, 5–8).
OCP/MCTD: Several UI-snRNP-derived peptide epitopes were shown to bind to OCP-associated HLA-DQ7 (DQB1*0301) and HLA-DR4 (DRB1*0401) molecules. Thus, three peptides bound to HLA-DQ7 (33%–26% optimal binding), and six epitopes bound to HLA-DR4 molecules (29%–15% optimal binding) (Supplemental Table 9A, B). On the other hand, 30 OCP-derived alpha 1 type XVII collagen peptides bound to MCTD-associated HLA-DR1 (DRB1*0101) (40%–17% optimal binding) and nine alpha 1 type XVII collagen peptides bound to the 9mer-HLA-B07 (59%–45% optimal binding). In the case of β4 integrin, 29 binders were detected for MCTD-associated HLA-DR1 (DRB1*0101) (37%–17% optimal binding) and five peptides were predicted to bind to the 9mer-HLA-B07 (62%–46% optimal binding). Peptide sequences are shown in Supplemental Table 9B.
MMP/MCTD
Analysis of MMP-specific β4 integrin binding to MMP-associated HLA-DQ7 (DQB1*0301) molecules, as well as the binding of MCTD-related UI-snRNP peptides to MCTD-specific HLA-DR1 (DRB1*0101) and the 9mer-HLA-B07 has been described in the previous sections of this report (Supplemental Tables 1, 5–9).
MMP/MCTD: In the dual disease model, the binding of β4 integrin to HLA-DR1 (DRB1*0101) resulted in 29 predicted epitopes (37%–17% optimal binding), while only five β4 integrin-derived peptides were found to bind to the 9mer-HLA-B07 (62%–46% optimal binding), as shown in Supplemental Table 10A. On the other hand, three UI-snRNP-derived peptides bound to HLA-DQ7 (DQB1*0301) molecules (33%–26% optimal binding). For peptide sequences refer to Supplemental Table 10B. Thus, based on similar values for peptide binding in these two disease models, it is possible that autoantigen-derived epitopes from one disease interact with the HLA alleles of the second disease.
BP/DH
Analysis of BP-specific autoantigens alpha 1 type XVII collagen and dystonin, as well as of DH-related TGM3 binding to HLA molecules known to be associated with BP (within a known disease model) has been performed and described in the previous sections of this study (Supplemental Tables 2, 4, 7 and 8).
BP/DH: Binding of type XVII collagen-derived peptides to DH-associated HLA-DQ8 (DQA1*0301/DQB1*0302) resulted in eight top binders (36%–25% optimal binding), and in one binder for HLA-DQ2 (DQA1*0501/DQB1*0201) molecules (optimal binding 45.89%) (Table 11A). For dystonin-related peptides, 12 were found to bind HLA-DQ8 (DQA1*0301/DQB1*0302) (47%–25% optimal binding) but none to HLA-DQ2 (DQA1*0501/DQB1*0201) molecules (Supplemental Table 11A). In the opposite direction, six TGM3-derived peptides were predicted to bind BP-associated HLA-DQ7 (DQB1*0301) molecules (45%–26% optimal binding). Peptide sequences are shown in Supplemental Table 11B.
Based on the predicted optimal binding data, these results suggest that epitopes derived from the autoantigenic proteins specific for one disease may bind to one or more HLA alleles associated with the second disease with various affinities. Most importantly, these observations show that while one peptide is capable of binding to its disease-associated HLA, a different peptide of the same autoantigen may bind to the HLA related to the second disease. This phenomenon may produce T cell recognition with different signaling outcomes, leading to the production of several autoantibody specificities characteristic for each of the induced disease conditions.
Discussion
It has been well documented that autoimmune diseases may coexist in the same patient, either sequentially or concurrently.50–64 PV, DH, BP, and SLE have all been reported in association with other autoimmune diseases as well as with each other. In particular, observations of dual autoimmunity in some patients who concurrently develop organ-specific and systemic disease have been reported.52,54,55,58,60,62,63 Multiple factors, including those of immunological, genetic, endocrine and environmental origin, contribute to the above condition. The immunogenetic mechanisms of this phenomenon present an intriguing unresolved problem of autoimmune predisposition, calling for development of prospective approaches of prediction and ultimately prevention of the disease. As a matter of fact, the involvement of T cells in immunopathogenesis of MCTD, PV and MMP has been well established. In MCTD, the role of anti-RNP-reactive T cells in autoantibody production has been demonstrated.65,66 In PV, it has been shown that B cells function as antigen-presenting cells stimulating Dsg3-specific CD4+ T helper (Th) cells to secrete cytokines such as interleukin (IL)-4, IL-6 and IL-10 which are required for proliferation of memory B cells and differentiation into antibody-producing plasma cells.67–69 Thus, the interplay between B and T cells seems to be critical, which is further supported by the finding that depletion of CD4+ T cells prevents antibody production. 69 Moreover, a clinical study showed that the mean frequency of Th2 CD4+ T cells was significantly elevated in PV patients with active disease, while no responses were detected for patients with disease in remission or controls. 71 Characterization of autoreactive T-cells has led to identification of immunodominant T-cell epitopes and the repertoire of Dsg3- or Dsg1-specific T-cells at the clonal level.72,73 Lastly, the potential role for antigen-specific autoreactive T cells in the pathogenesis of MMP has also been addressed.74,75 Collectively, these observations suggest that T cell epitopes of the respective autoantigens may bind to their HLA molecules and trigger the activation of autoreactive T cells, which in turn would induce production of pathogenic autoantibodies.
In the present study, a bioinformatics-based search for potential epitopes restricted by HLA molecules associated with autoimmune mucocutaneous blistering diseases and systemic diseases has been undertaken in order to address the immunogenetics of the phenomenon of shared autoimmunity. The data across different disease combinations obtained in this report suggest that coexistence of two autoimmune diseases in the same patient might be triggered by the following mechanisms: 1) binding of each of the autoantigenic peptides related to a specific disease, to its specific HLA-associated molecules, or 2) binding of two epitopes derived from the same autoantigen to two HLA class II molecules, each associated with one disease. This binding may potentially result in differential signaling leading to the generation of disease-specific antibodies which contribute to the pathogenesis of both disease conditions. The data on the number of predicted peptide binding epitopes for each disease combination described in this study is presented in Table 2. The highest number of binders was found for OCP while the lowest was in the case of DH, BP/MCTD and MMP/MCTD. Figure 1 shows disease models arranged by the number of predicted binders from other diseases. In each case, the point of reference is the HLA class II molecule. An analysis of in-model binding between HLA II molecules and associated protein within a given disease model is compared to the binding predictions for those same HLA II molecules against the set of proteins associated with the second disease model (cross-model analysis).
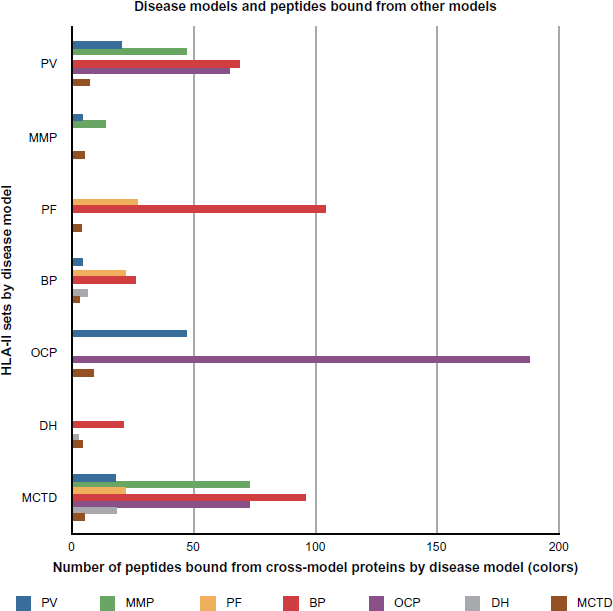
Disease models and peptides bound from other models.
It should be noted that there are certain limitations to the methodology employed in this study. The binding matrices used are based on motifs developed from sequences known to bind to the MHC for which the matrix is developed. There are inherent problems which limit the suitability of the matrices for prediction of novel binding sequences, particularly the variable number of known binding sequences available for the development of binding matrices, in some cases quite small. Another limitation is that laboratory confirmation of binding predictions has yet to be completed. However, a recent publication has identified a peptide epitope derived from BP180 associated with IgA dermatosis that was shown to interact with monoclonal antibodies
76
. Parts of BP180 mapped sequences SMDR
Following pioneering biochemical studies which led to elucidation of peptide motifs associated with class I and class II MHC receptors,77–80 numerous analyses on the nature of the peptide/MHC interactions in the context of autoimmune/inflammatory diseases have been performed showing promiscuity of peptide binding to class II MHC molecules.81,82 In the present study, each autoantigen of interest was subjected to the cross-HLA II-binding analyzer tool resulting in multiple peptides which were ranked based on the probability of their binding to the respective HLA molecule. It is of interest that, in spite of the large number of sequences generated for each autoantigen (varying from 429 for UI-snRNP to 2641 for dystonin) the number of the actual binders above the threshold was specific for each peptide/HLA combination and resulted in either none, very few or multiple binders, as shown in Supplemental Tables 1–11. This is likely due to the polymorphism in the HLA pocket residues allowing specific peptide motifs to bind based on the size and hydrophobicity of the pockets, so that only the matching amino acids of the peptide would fit in.
Sequences of autoantigenic immunodominant epitopes for a number of blistering diseases have been previously reported.83–93 In the most studied model, PV, extensive analysis of peptide motifs bound to the susceptible alleles showed sequence specificities and variability in the HLA binding pockets.84–86 For example, at least nine previously identified stimulatory Dsg3 peptide sequences corresponding to the amino acid residues 96–112 (PFGIFVVDKNTGDINIT), 191–205 (NSKIAFKIVSQEPAG), 206–220 (TPMFLLSRNTGEVRT), 252–266 (ECNIKVKDVNDNFPM), 342–356 (SVKLSIAVKNKAEFH), 380–394 (GIAFRPASKTFTVQK), 763–777 (SGTMRTRHSTGGTNK), 810–824 (NDCLLIYDNEGADAT) and 963–977 (ERVICPISSVPGNLA) were shown to bind to PV-associated HLA-DR4 (DRB1*0402) and DQ5 (DQB1*0503) with a sliding window of up to three amino acids for the core residues.
84
In the present study, our binding analysis of Dsg3 epitope binding to DRB1*0402 revealed several sequences that were identical to the reported core and flanking residues within the peptide Dsg3 (flanking residues spaced, core underlined): NSK
It is of interest to note that, according to Mouquet et al, 87 human Dsg1 was found to contain a T cell epitope capable of binding to PF-associated HLA Class II DRB1*0102 molecules. In contrast, no predicted binders derived from Dsg1 were detected for HLA-DR1 (DRB1*0102) in the present study (Supplemental Table 2). Also of interest, a recent study showed that the HLA-DR3 (DRB1*0301) molecule is linked to endemic PF in Tunisian patients. 92 It would be of interest to analyze the peptide binding predictions on Dsg1 to this PF-associated HLA molecule in order to identify potential epitopes.
Importantly, no similar sequences were found in two different autoantigens specific for the two diseases, suggesting that promiscuous binding of the same epitope to the two HLA alleles associated with these diseases is unlikely to be the cause of shared autoimmunity. Rather, the data presented in this study demonstrates the possibility that an additional epitope derived from the same autoantigen binds to the HLA specific for the second disease. To our knowledge, this is the first study suggesting the potential mechanism of the induction of dual autoimmunity mediated by epitopes derived from a single autoantigen. In support of this mechanism, it is noteworthy that the affinity of the binding between the autoantigenic peptide epitope, the susceptible HLA and the TCR play an important role in T cell activation. Due to certain degree of promiscuity and specificity in peptide recognition by the HLA receptors, not a single binding affinity, but rather a range of affinities would account for the productive interaction between the peptide epitopes, HLA and the TCR, leading to T cell-mediated B cell activation and antibody secretion. Modeling of the bound conformation of PV-associated peptides revealed the role of DRB1*0402 in the selection of specific self-epitopes. 84 Several studies suggest that autoantigenic peptides do not necessarily bind to disease-associated HLA molecules with high affinity, but rather within the intermediate range, thus allowing for the rescue of autoreactive T cells by virtue of weaker HLA/peptide/TCR interactions. In contrast, protective HLA proteins are more efficient binders of self-antigens, which results in elimination of autoreactive T cells. 85
It should be noted that peptide binding to HLA is facilitated by the interactions between the amino acid residues lining the groove of HLA molecules and the side chains of the bound peptide. The binding pockets of HLA class II, defined by the polymorphic β chain and the more conservative α chain of the αβ heterodimer, share homology between some alleles but may also differ from other alleles as defined by size, charge and hydrophobicity. 83 Thus, autoantigens may not share sequence homology, but still may encompass peptides able to bind different HLA due to the presence of certain amino acids which would fit to the binding pockets of the HLA molecules. In spite of this fact, the two peptides may share common binding motifs, dictated by structural requirements of the HLA pockets accommodating the peptides. Due to the degenerate nature of the HLA binding and TCR recognition, an observation which has been widely accepted for the past decade, 94 common binding motifs would be sufficient to allow peptide binding to different HLA molecules. However, in this case, it is possible that the recognition of the HLA/peptide complex by T cells will differ depending on the orientation of the TCR interacting with the amino acids facing away from the binding groove, and thus will result in differential activation by T cells.
Molecular and cellular mechanisms governing the concurrent or sequential presence of autoimmune blistering and systemic diseases in patients remain to be elucidated. Investigation of these mechanisms has been significantly delayed due to the lack of animal models in which both the systemic and organ-specific autoimmune diseases can be induced. To this end, only a small number of experimental models of susceptibility to a single disease have been developed with limited success.95–97 Development of such animal models allowing investigation of the effects of the triggering factors on shared autoimmunity would require genetic manipulations enabling the introduction of elements of susceptibility, ie, human HLA and/or autoantigen-specific TCR/BCR. Thus, a transgenic mouse model expressing two disease-associated HLA and two TCR/BCR specific for each of the autoantigenic peptides would be most suitable for this purpose. In these mice, the experimental approach would include the administration of disease-inducing peptides, separately or concomitantly, and monitoring the animals for manifestations of each disease. In parallel, ex-vivo functional analysis including antigen-specific proliferation, cytokine secretion and antibody phenotyping, has to be performed. The in vitro binding studies employing purified HLA proteins and synthetic peptides, and the cellular assays with antigen-presenting cells and patient's lymphocytes would also be instrumental.
To our knowledge, this is the first study reporting extensive analysis of peptide binding predictions to a number of HLA alleles associated with autoimmune blistering and systemic diseases. Further studies of these patients, and especially of T and B lymphocytes administered into HLA-transgenic mice, will provide valuable information on cellular and molecular mechanisms critical for immunoregulation and production of pathogenic autoantibodies. Such studies have significant clinical ramifications and implications for the development of novel immune therapies targeting both autoimmune diseases. The elucidation of HLA-restricted immune recognition mechanisms prompting the production of two or more disease-specific autoantibodies holds significant clinical ramifications and implications for the development of more effective treatment protocols. Currently, blistering and systemic diseases are treated by a number of protocols, including administration of IV Ig regiments, bone marrow transplantation, steroids (prednisone, prednisolone, dapsone, clobetasol), adjuvant drugs (azathioprine, mycophenolate mofetil, cyclosporine, rituximab) and emerging treatments by gene therapy or stem cell transplantation. These treatments are aimed at suppression or replacement of affected proteins and cells, with no specificity of targeting individual components.
Our findings provide important information on the identity of potential epitopes implicated in pathogenesis of blistering and systemic diseases, and specifically, on autoantibody reactivity in these patients. Potential therapies for these conditions could include targeted strategies to eliminate these autoantibodies and/or combination therapy with agents directed against several such specificities.
Abbreviations
AMBD, autoimmune mucocutaneous blistering diseases; APC, antigen-presenting cells; BP, bullous pemphigoid; DH, dermatitis herpetiformis; Dsg, desmoglein; HLA, human leukocyte antigens; IL, interleukin; MCTD, mixed connective tissue disease; MMP, mucous membrane pemphigoid; OCP, ocular cicatricial pemphigoid; PF, pemphigus foliaceus; PV, pemphigus vulgaris; RNP, ribonucleoprotein antigen; TCR, T cell receptor; Th, T helper cells.
Disclosure
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
Supplementary Material
Peptide sequences of BP- and MCTD-derived autoantigens predicted to bind their HLA-associated alleles in a one- and two-disease model.

| Disease | MHC | Threshold | Autoantigen | Total predicted binders | Top binders | Score | % optimal |
|---|---|---|---|---|---|---|---|
| BP-BP | HLA-DQ7 (DQB1*0301) | 11.701 | alpha 1 type XVII collagen (BP180) | 14 | IRRSILPYG | 19.68 | 43.09 |
| FDYSELASH | 19.381 | 42.44 | |||||
| STDASHSRG | 16.697 | 36.56 | |||||
| ILDANLPSH | 16.398 | 35.9 | |||||
| AGPAGLPGH | 16.234 | 35.55 | |||||
| VWSSTLPAG | 15.634 | 34.23 | |||||
| SLGAGGAFG | 13.849 | 30.32 | |||||
| IRGPPGPSG | 13.625 | 29.83 | |||||
| SSQSVSGTY | 13.531 | 29.63 | |||||
| NTNAYSAGS | 13.391 | 29.32 | |||||
| YRRAHSPAS | 12,928 | 28.31 | |||||
| LSSYLHTAG | 12,403 | 27.16 | |||||
| DIHSYGSSG | 12,396 | 27.14 | |||||
| APGPAGPAG | 11,938 | 26.14 | |||||
| dystonin (BP230) | 12 | FESYGHSSH | 16,983 | 37.19 | |||
| NFDGDHACS | 15.913 | 34.84 | |||||
| YRDTYHPLD | 14,818 | 32.45 | |||||
| LTPSVTPAY | 14,805 | 32.42 | |||||
| IEPQVHSRL | 14,386 | 31.5 | |||||
| FAQTLHPSL | 14,356 | 31.43 | |||||
| ITQSLNSGF | 12,997 | 28.46 | |||||
| LLQRQKATV | 12,573 | 27.53 | |||||
| LRHTVTARQ | 12,495 | 27.36 | |||||
| ADFDFHTGL | 12,406 | 27.16 | |||||
| ISPTGNEAM | 12.06 | 26.41 | |||||
| IIDVLIATK | 12,013 | 26.3 | |||||
| BP-MCTD | HLA-DQ7 (DQB1*0301) | 11.701 | transglutaminase 3 | 6 | GSDSVWNFH | 20,579 | 45.06 |
| DDNGVLAGN | 17,095 | 37.43 | |||||
| YVGRVLSAM | 14,523 | 31.8 | |||||
| DPRSWNGSV | 13.59 | 29.76 | |||||
| AEHPIKISY | 13,343 | 29.22 | |||||
| DH-DH | HLA-DQ2 (DQA1*0501/DQB1*0201) | 31.451 | transglutaminase 3 | 0 | ITAVCKVPD | 11.92 | 26.1 |
| HLA-DQ8 (DQA1*0301/DQB1*0302) | 12.635 | transglutaminase 3 | 3 | DFSCNKFPA | 15.737 | 30.63 | |
| ALRSLGIPS | 14.828 | 28.86 | |||||
| SATMSLDPE | 14.644 | 28.5 | |||||
| DH-MCTD | HLA-DQ2 (DQA1*0501/ DQB1*0201) | 31.451 | alpha 1 type XVII collagen (BP180) | 1 | RGREGPMGP | 32.97 | 45.89 |
| 31.451 | dystonin (BP230) | 0 | |||||
| HLA-DQ8 (DQA1*0301/DQB1*0302) | 12.635 | alpha 1 type XVII collagen (BP180) | 8 |
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|||||
| DRGPAGPPG | 15,018 | 29.23 | |||||
| WGPAPAWCP | 14,162 | 27.56 | |||||
| PKGDRGFPG | 13,865 | 26.99 | |||||
| DRLQGMAPA | 13,846 | 26.95 | |||||
| GAKGAMGPA | 13.11 | 25.52 | |||||
| 12.635 | dystonin (BP230) | 12 | ITRAHAVAE | 24,137 | 46.98 | ||
| IKRCKETSE | 20,169 | 39.26 | |||||
| PAYTPGFPS | 18,052 | 35.14 | |||||
| VSWHYLINE | 15,507 | 30.18 | |||||
| VQRVAKLRD | 15,481 | 30.13 | |||||
| AYRAAMQTQ | 15,471 | 30.11 | |||||
| DEIMALRNE | 15,444 | 30.06 | |||||
| VRGIRVPPE | 15,033 | 29.26 | |||||
| IVREKEAAE | 14,276 | 27.79 | |||||
| VLKGVVDPE | 13,926 | 27.11 |