
Abstract
Malaria is a complex parasitic disease that is currently causing great concerns globally owing to the resistance to antimalarial drugs and lack of an effective vaccine. The present study involves the characterization of extracellular secretory proteins as vaccine candidates derived from proteome analysis of
Introduction
Malaria is caused by five species of protozoan parasites that affect humans, belonging to the genus
The asexual blood stage of the parasite is responsible for this disease symptom and, consequently, has been the focus of research for vaccine development that imparts long-term immunological memory.
2
This memory is a cardinal property of the adaptive immune system where memory T- and B-cells play a crucial role in providing long-term protective immunity.3,4 Now it is widely accepted that B-cells and CD4+ T-cells are activated during the blood stages and CD8+ T-cells are activated during the pre-erythrocytic or liver stage of the
Therefore, it is important to understand the response of CD8+ and CD4+ T-cell epitopes induced by blood-stage infection, which may prove to be essential in the development of a vaccine that targets the erythrocytic stage of the malaria parasite. In order to design an effective vaccine leading to the generation of memory T- and B-cells against malaria, a sound understanding of the quality and quantity of the immune responses generated against malarial infection at the blood stage seems imperative. 9 Since humoral and cellular immune responses are interrelated, a functional B-cell response is not generated in the absence of T-cell help, and vice versa. However, to date, most vaccine formulations prepared against malaria have targeted only the induction of an antibody response. Because of the complex life-cycle of the parasite and its antigenic make-up, a vaccine design strategy that also incorporates T-cell-mediated immunity has its own benefits. 10 Thus, the T-cell epitopes that promiscuously bind multiple alleles of a human leukocyte antigen (HLA) may be prime targets for future vaccine and immunotherapy development because they seem relevant to a large proportion of the human population. 11
However, the experimental determination of binding specificities for even a single antigen and a single MHC allele is an expensive, laborious, and time-consuming process; at the same time, it is not practical to undertake a binding study of the MHC supertypes, which will involve large numbers of alleles.
12
Hence, the use of immunoinformatics tools for the initial screening and designing of suitable vaccine candidates is desirable covering antigenic variability and human MHC allelic diversity.13,14 Keeping the above facts in mind, the present study reports the use of a novel proteome mining strategy that integrates bioinformatics-based antigen/epitope predictions and HLA-supertype considerations for the purpose of identifying potentially immunogenic proteins from the asexual blood stage of
Methods
Retrieval of proteome sequence dataset
The proteome analysis of
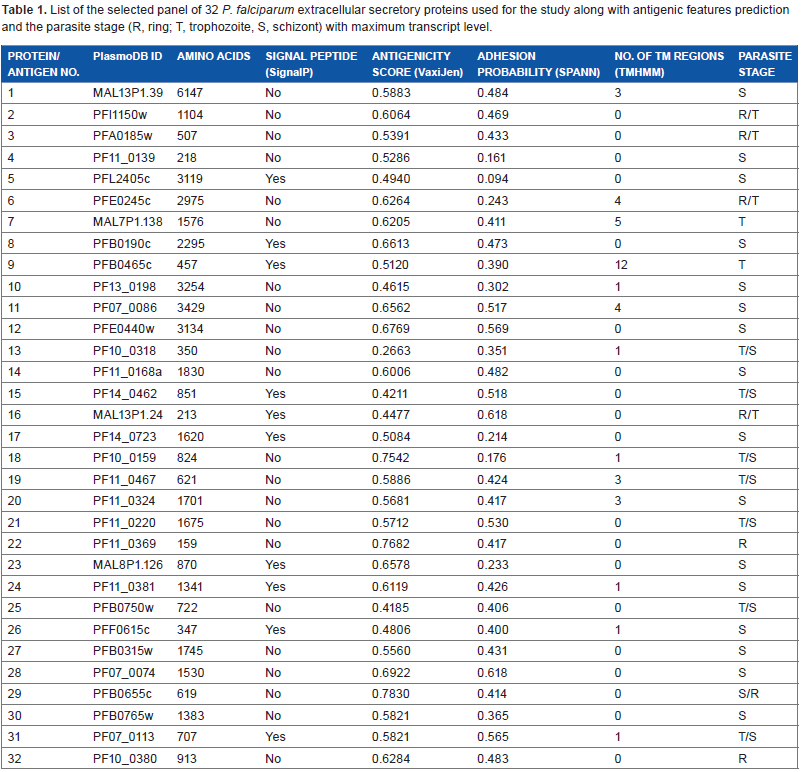
List of the selected panel of 32
Methodology of the work
The characterization of potential vaccine candidates of the selected 32 proteins was done using the following computational tools: VaxiJen,17,18 SPAAN,
19
Target P,
20
SignalP,
21
TMHMM,
22
and Allermatch™.
23
Moreover, similarity to human and mouse reference proteins and other related
In order to identify the immunogenic proteins, the following immunoinformatics tools were used. MULTIPRED2 (http://cvc.dfci.harvard.edu/multipred2/) is a computational system for large-scale screening of peptide binding to multiple alleles belonging to HLA class I (-A, -B and -C) and class II (-DR) supertypes as well as to alleles belonging to an individual's genotype using netMHCpan 2.0 25 and net-MHCIIpan 1.0 26 algorithms, respectively. Peptides with predicted half-maximal inhibitory concentration (IC50) ≤500 nmol/L (nM) and >50 nM are considered as weak binders and those ≤50 nM as strong binders. NetCTL (http://www.cbs.dtu.dk/services/NetCTL) was used for the prediction of naturally processed CD8+ T-cell epitopes, integrating predictions of MHC class I binding affinity, TAP transport efficiency, and C-terminal proteasomal cleavage analysis. It has been demonstrated to have higher predictive performance than other similar Web servers such as EpiJen, MAPPP, MHC-pathway, and WAPP on all performance measures. 27 For predicting CD8+ T-cell epitopes, peptides binding to HLA class I supertypes with IC50 (binding affinity) ≤500 nM, equivalent to the transformed binding affinity value ≥0.426 [ie, 1–-log 50 k (aff nM)], the weight on C-terminal cleavage 0.1, and on TAP transport 0.05 were used, which resulted in optimal predictive performance. Peptides with a combined prediction threshold score ≥0.75 were marked as potential CD8+ T-cell epitopes, as suggested by Larsen et al. 28 The methodology used for computational antigen characterization and T-cell epitopes analysis is given in Figure 1.
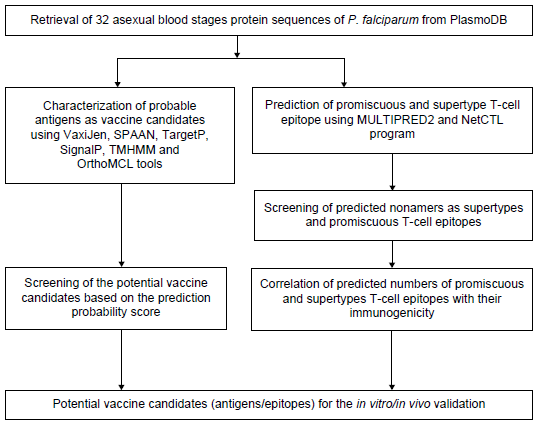
Flow chart showing the methodology used for the prediction of antigen characteristics and immunogenicity analysis using reverse vaccinology approach.
Results and Discussion
Current development of subunit vaccines are based on a single or a few antigens, and may therefore elicit too narrow response, providing suboptimal protection, further worsened by genetic diversity.
29
Doolan et al
15
and Crompton et al
10
have demonstrated that immunization with irradiated sporozoites induced protective immune responses that were broadly dispersed not only on a relatively large number of parasite antigens but also against multiple epitopes on those antigens with variable potency. Therefore, a robust approach to diverse antigen selection can be employed with multiple antigenic traits encompassing antigen structure, location, function, and results from various assays and models.
30
The genomes of the
Vaccine candidate characterization
In order to characterize the panel of 32 asexual blood-stage proteins as vaccine candidates, the antigenic feature prediction tools available in the public domain were used. Since adhesins (surface proteins) are required at various stages of the malaria parasite life-cycle and often considered as good vaccine targets, the prediction for being an adhesin and adhesin-like was done using the SPANN program30 on the basis of their adhesin probability threshold score above 0.51. Out of the 32 proteins, 8 (nos. 11, 15, 21, 31, 12, 41, 16, 28) were predicted as adhesins (Table 1). Furthermore, surface-exposed proteins such as outer membrane proteins and secreted proteins are usually ideal targets for vaccine development. Hence, the localization prediction of a secretory pathway was done through the TargetP 20 and SignalP 21 Web servers, which revealed important understanding of its site of action and functionality as a surface protein. Only 10 proteins (nos. 5, 8, 9, 15, 16, 17, 23, 24, 26, 31) cleared the test of SignalP; however, only 5 proteins (nos. 5, 17, 23, 24, 26) were predicted to contain secretory signal peptide using TargetP (Table 1).
Since it is difficult to clone, express, and purify proteins with more than one TM spanning region, such regions were predicted using the TMHMM
22
Web server. It was found that 26 (except nos. 6, 7, 9, 11, 19, 20) out of the 32 proteins had less than two TM spanning regions (Table 1). Since orthologous information for a vaccine candidate gives an idea of the probable use of the vaccine in other related species, an exhaustive search for potential orthologs was performed using a homology prediction program through the OrthoMCL database.
24
Most of the selected proteins have orthologs in the rodent malarial genomes (
Most of the malaria vaccines work mainly by inducing serum antibodies, a necessary and often sufficient component of vaccine efficacy; however, it is well known that CD8+ T-cell responses are a key element of the immune reactivity elicited by several
T-cell epitopes analysis of asexual blood-stage proteins
Recent approach to identify targets of CD8+ and CD4+ T-cell responses in an antigen is based on the prediction of high-affinity binding class I or class II restricted T-cell epitopes using computational algorithms. These predicted peptides bind specific common HLA types or multiple alleles within an HLA supertype, and thereby provide coverage representative of all populations with ethnic freedom.41–43 As suggested by Doolan et al,
15
this strategy would lead to a “hit” even if the accuracy of the predictions is as low as 10%. Scientific literature also depicted a correlation between immunogenicity and the capacity to bind multiple molecules from a given HLA supertype.44–46 The high-affinity binding to prototype allele of a given supertype has been shown to be highly predictive of supertype binding and is more immunogenic
Analysis of HLA class I and II supertypes restricted epitopes
A comprehensive analysis of predicted HLA class I supertype epitope distribution [HLA-A (A1, A2, A3, A24, A26), HLA-B (B7, B8, B27, B44, B58, -B62), HLA-C (C1, C4)] is shown in Figure 2. A total of 41,090 probable CD8+ T-cell epitopes were predicted, including 20,025 HLA-A, 20,731 HLA-B, and 334 HLA-C supertype binding peptides. Further, according to the MULTIPRED2 algorithm, peptides that were predicted to bind at least 50% of the alleles in a supertype are considered as promiscuous binders. The percentage of promiscuity was calculated using the number of alleles predicted to bind the peptide divided by total number of alleles in a supertype. Thus, a total of 4,055 promiscuous CD8+ T-cell epitopes were predicted including 2,343 in HLA-A and 1,712 in HLA-B supertypes (Fig. 3). No promiscuous epitopes were predicted for the HLA-C supertype. Promiscuous T-cell epitopes are advantageous for vaccine design because they effectively increase the number of epitopes to which an individual can respond and provide much more extensive coverage of the population.
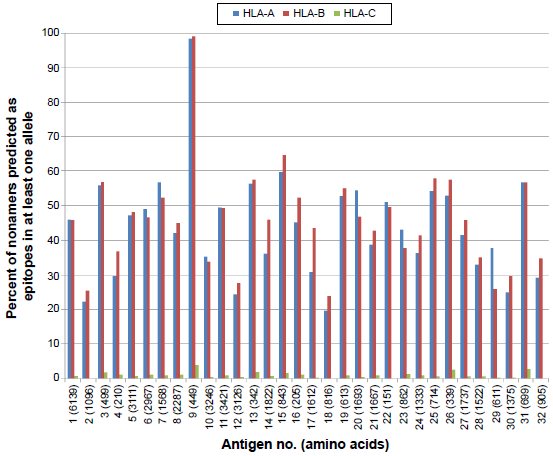
Percent of nonamers predicted as epitopes of HLA-A (A1, A2, A3, A24, A26), HLA-B (B7, B8, B27, B44, B58) and HLA-C (C1, C4) supertypes binding with at least one HLA allele in the supertype against the selected panel of 32
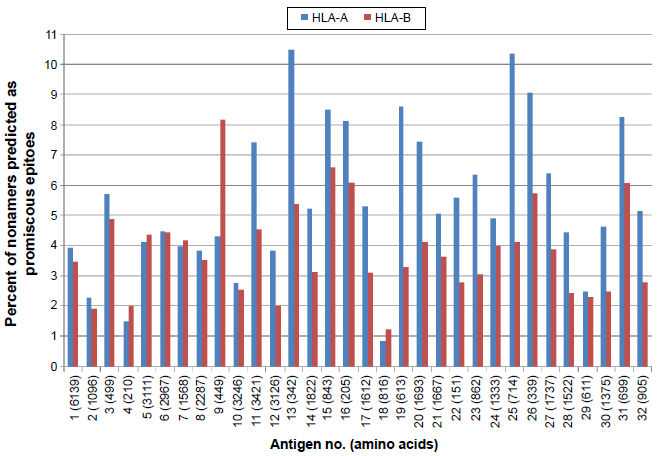
Percent of nonamers predicted as promiscous epitopes of HLA-A and HLA-B supertypes which binds with at least 50% of HLA alleles in the supertype (A1, A2, A3, A24, A26, B7, B8, B27, B44, B58) against the selected panel of 32
A similar comprehensive analysis of MULTIPRED2-predicted HLA class II-DR (DR1, DR3, DR4, DR6, DR7, DR8, DR9, DR11, DR12, DR13, DR14, DR15, and DR16) supertype epitope distribution was also done against the selected 32 proteins. A total of 2,657 probable CD4+ T-cell epitope candidates were predicted including 689 DR1, 327 DR3, 312 DR4, 137 DR6, 289 DR7, 28 DR8, 122 DR9, 112 DR11, 119 DR12, 159 DR13, 119 DR14, 175 DR15, and 70 DR16 supertype binding peptides (Fig. 4). Out of 2,657, a total of 845 promiscuous CD4+ T-cell epitopes were screened using the MULTIPRED2 algorithm (Fig. 5). On the basis of the percent of nonamers predicted as HLA class I and II supertypes epitopes, protein antigens are grouped into four major categories: 12 highly immunogenic antigens (nos. 3, 7, 9, 13, 15, 16, 19, 20, 22, 25, 26, 31) having ≥50% of nonamers predicted as supertype epitopes; 11 intermediately immunogenic antigens (nos. 1, 5, 6, 8, 11, 14, 17, 21, 23, 24, 27) having ≥40% and <50% of the predicted epitopes; 5 weakly immunogenic antigens (nos. 4, 10, 28, 29, 32) having <40% and ≥30% of the predicted epitopes; and remaining antigens characterized as poorly immunogenic (Figs. 2–5). Finally, 18 antigens (nos. 3, 9, 11, 13, 14, 15, 16, 17, 19, 20, 21, 22, 23, 25, 26, 27, 31, 32) that are either highly or intermediately immunogenic have more than 5% of nonamers predicted as promiscuous epitopes of HLA-A, -B, or -DR supertypes (Figs. 3 and 5). These prediction data are useful for novel malaria vaccine design because the promiscuous and supertype peptide binders enable the reduction of the number of predicted epitopes without compromising the population coverage.50,51
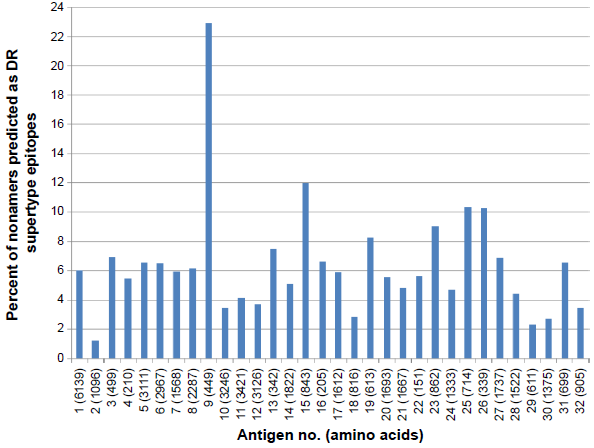
Percent of nonamers predicted as epitopes of HLA-DR supertypes which binds with at least one HLA alleles in the supertype against the selected panel of 32
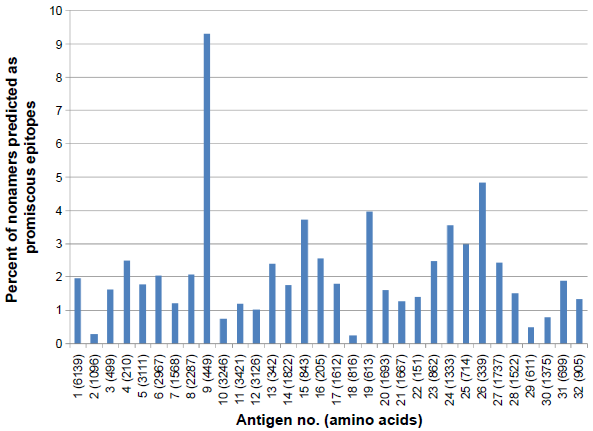
Percent of nonamers predicted as promiscous epitopes of HLA-DR supertypes which bind with at least 50% of HLA alleles against the selected panel of 32
Further, it is important to consider whether each MHC binding peptide is being correctly processed from the native antigen and subsequently displayed on the surface of antigen-presenting cells. At present, it is possible to predict the naturally processed peptides using the NetCTL algorithm with a combined epitope prediction score of 0.75, which includes predictions of proteasomal cleavage, TAP binding, and HLA binding. The performance of the integrated method is found to be significantly higher than that of the MHC binding prediction methods. In practical terms, the experimental effort needed to identify an epitope in a hypothetical protein with 85% probability is reduced by 20–30% when using the integrated method. 28 Thus, NetCTL can be used to effectively predict the pools of supertype epitopes and immunogenicity of a novel protein. 52
Analysis of naturally processed HLA class I supertypes restricted epitopes
A comprehensive analysis of epitope binding prediction was done using NetCTL against the HLA-A (A1, -A2, -A3, -A24, -A26) and HLA-B (B7, -B8, -B27, -B39, -B44, -B58, -B62) supertypes for the selected 32 proteins. A total of 17,098 supertype CD8+ T-cell epitopes were predicted, including 9,028 HLA-A and 8,070 HLA-B supertypes (Fig. 6).
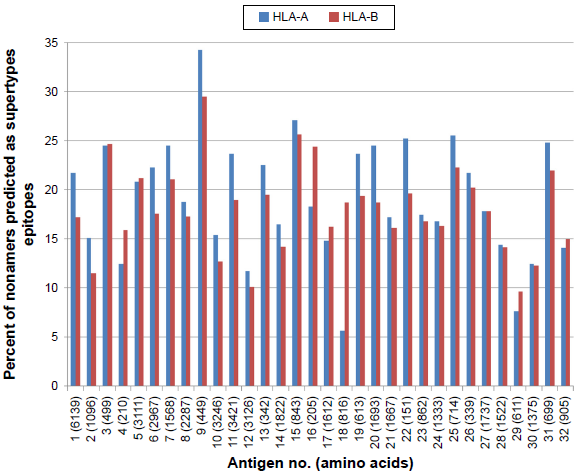
Percent of nonamers predicted as epitopes of HLA-A (A1, A2, A3, A24, A26) and HLA-B (B7, B8, B27, B39, B44, B58, B62) supertypes against the selected panel of 32
Similarly, based on the percent of nonamers predicted as naturally processed supertype epitopes, 32 proteins are grouped into four major categories: 16 highly immunogenic antigens (nos. 1, 3, 5, 6, 7, 9, 11, 13, 15, 16, 19, 20, 22, 25, 26, 31) having more than 20% predicted supertypes epitopes; 11 intermediately immunogenic antigens (nos. 2, 4, 8, 10, 14, 17, 18, 21, 23, 24, 27) having >15% and ≤20% predicted supertype epitopes; 4 weekly immunogenic antigens (nos. 12, 28, 30, 32) with >10% and ≤15% predicted supertypes epitopes; and 1 poorly immunogenic antigen (no. 29) having <10% predicted supertypes.15,51 On comparing the cost and time for
Furthermore, in the study of Doolan et al,
15
it has been shown that antigenicity against the
The data presented herein may have several important implications. First, a strategy to mine proteomic data for the identification of novel CD4+ and CD8+ T-cell epitopes recognized by multiple genetic restrictions has been described. Second, these data also suggest that strict immunodominance (ie, only one or a few antigens or epitopes dominating the responses) does not apply in the context of a complex parasite like
Conclusion
Currently, there is a lot of concern regarding the designing an effective malaria vaccine for a global population. The findings reported here suggest new ways forward for omics-guided vaccine target discovery, building on a reverse vaccinology approach. In the selected 32 proteins, of which 27 were novel extracellular antigens, half of the antigens are either highly or intermediately immunogenic, consensually predicted by NetCTL and MULTIPRED2 programs. Notably, we found that most of the selected proteins contain at least 10% of nonamers as HLA-A and -B restricted supertype CD8+ T-cell epitopes. We anticipates that T-cell immune responses against asexual blood stages of
Author Contributions
Conceived and designed the experiments: SPS. Analyzed the data: VV. Wrote the first draft of the manuscript: SPS, VV. Contributed to the writing of the manuscript: BNM. Agreed with the manuscript, results, and conclusions: BNM. Jointly developed the structure and arguments for the paper: SPS, BNM. Made critical revisions and approved the final version: SPS. All authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgments
The authors are grateful to Amity University Uttar Pradesh, Lucknow Campus and the U.P. Technical University for providing the necessary infrastructure and support for this work. They are also thankful to Dr. Brijesh Pandey and Dr. A.K Singh, Amity University Uttar Pradesh, Lucknow Campus for valuable suggestions.