
Abstract
It is now broadly accepted that white adipose tissue disorders, such as obesity, are associated with a chronic low-grade inflammation predisposing to the development of insulin-resistance, type 2 diabetes and cardiovascular complications. In obesity, accumulation of visceral adipose tissue, rather than subcutaneous adipose tissue, is regarded as the most critical factor contributing to the pathogenesis of these metabolic diseases. Recently has emerged the notion that inflammatory response accompanying obesity corresponds to a cytokine-mediated activation of innate immunity.
The purpose of this review is to provide an update on this emerging concept and to show the reader how innate immune metabolic pathways engaged within white adipose tissue could interfere with innate inflammatory immune defense. First, adipose tissue is reported as an important in vivo source of inflammatory cytokines and adipocytes express some receptors of the innate immune system (namely the Toll-like receptors). Second, both innate and adaptive immune cells (respectively, macrophages, dendritic-like cells and T-lymphocytes) appear more and more essential to the initiation and the development of adipose tissue inflammation. More specifically, adipose tissue macrophages have recently emerged as key players in the inflammatory process of obese adipose tissue. Their number and their phenotypic switch from a non inflammatory (i.e. M2) to an inflammatory (i.e. M1) state are likely crucial in the onset of obese adipose tissue inflammation and in the development of insulin-resistance. Finally, the hormonal regulation of adipose tissue inflammation is exemplified by recent data regarding the role of glucocorticoids, both at the level of adipose cells and macrophages.
Altogether, adipose tissue might therefore be regarded as a true immune organ, at the crossroad between metabolism and immune system.
The Adipose Tissue: At the Crossroad between Innate and Adaptive Immune Systems
The apparent simplicity of white adipose tissue (WAT), histologically and metabolically, is the main reason why it has relatively been ignored for a long time. Indeed, the primary function of WAT is to store energy in the form of triglycerides during periods of energy excess and to release energy during fasting or starvation as free fatty acids and glycerol. This simplicity is, however, illusory. At the cellular level, there is some heterogeneity in white adipose tissue, with approximately one third of lipid-filled cells (i.e. mature adipocytes) whereas the remaining 2/3 is mostly composed of stromal vascular cells (i.e. fibroblasts and adipocyte precursors) associated with endothelial cells, nerves, and immune cells such as macrophages (cf
Thereby, adipose tissue has lately switched from being a passive and silent energy “reservoir” to representing a complex, highly active and essential metabolic and endocrine organ, secreting an assortment of hormones, cytokines, chemokines and growth factors which regulate whole-body metabolism and immune function.
Below, we will sum up some critical studies demonstrating that adipose tissue might be regarded as a new member of the immune system (cf I) and that the inflammatory process, which is an essential early event in the development of obesity, starts and develops within the adipose tissue thanks to the cells of both the innate and adaptive immune systems (cf
Adipose Tissue as a Source of Inflammatory Cytokines
As stated above, the identification of leptin in 1994 2 led to the recognition that WAT is an important endocrine secretory organ. Indeed, white adipocytes secrete a multiplicity of factors termed “adipokines”,highly diverse in terms of both structure and function (reviewed in 3 ). These factors encompass cytokines (e.g. TNFα, interleukin-6 (IL-6)), chemokines (e.g. monocyte chemoattractant protein-1 (MCP-1)), proteins of the alternative complement system (e.g. adipsin), and a series of proteins involved in processes ranging from regulation of blood pressure (e.g. angiotensinogen), to glucose homeostasis (e.g. leptin, adiponectin) and angiogenesis (e.g. vascular endothelial growth factor).
Importantly, several adipokines are linked to inflammation and immune response. 3 The inflammation-related adipokines include cytokines (e.g. TNFα, IL-1β, −6, −8, −10, −18, transforming growth factor β (TGFβ) and macrophage migration inhibitory factor), chemokines (MCP-1), and acute phase proteins. In addition, the two major adipocyte hormones, leptin and adiponectin, were shown to exert, respectively, pro- and anti-inflammatory actions. 4 6
TNFα and IL-6 are the best-studied adipocyte-derived pro-inflammatory factors, both are increased in adipose tissue with obesity.7,8 TNFα was the first inflammatory cytokine shown to be produced by adipocytes, 1 even though adipose tissue macrophages have been later identified as being the main cell source in this tissue 9 (cf II. l). TNFα level is increased in adipose tissue and plasma of obese patients and has been related to obesity-associated complications.7,8 The involvement of TNFα in insulin-resistance probably results from its multiple direct effects on adipocytes, ranging from alteration of adipocyte differentiation, metabolism, insulin sensitivity and endocrine function (reviewed in10,11). Indeed, TNFα inhibits the transcription of many mature adipocyte-specific genes, such as those involved in glucose uptake (e.g. glucose transporter-4), insulin responsiveness (e.g. insulin receptor and insulin receptor substrate-1), 12 14 and lipogenesis (e.g. lipoprotein lipase).15,14 NF-kB activation is necessary for TNFα-induced repression of many adipocyte-specific genes. 14 Importantly, the expression level of the nuclear factor peroxisome proliferator-activated receptor γ (PPAR γ), which is necessary to maintain mature adipocyte phenotype, is down-regulated by TNFα exposure. 14 TNFα also stimulates lipolysis 16 via various mechanisms. Overall, TNFα reduces adipocyte capacity for triglyceride storage and promotes adipocyte insulin resistance. Indeed, beside its impact on adipocyte gene transcription, TNFα has also been shown to negatively interfere with the insulin signaling pathway. 17 The cytokine also down-regulate the mRNA level of adiponectin,13,14 an adipocyte-derived hormone which contributes to the maintenance of peripheral glucose and lipid homeostasis. Moreover, TNFα inhibits the conversion of pre-adipocytes to mature adipocytes, allowing further recruitment of uncommitted cells and thus possible expansion of adipose tissue mass. 18 Nevertheless, its influence on immune response mostly results from its enhancing effect on the production of other cytokines, such as IL-6, rather than from a direct effect.
Like TNFα, the levels of the other major inflammatory cytokine IL-6 correlate with body mass index.8,19 One third of circulating IL-6 is produced by adipose tissue, with visceral WAT producing more IL-6 than subcutaneous WAT. In fat tissue, only a fraction (estimated to ~10%) of IL-6 is secreted by adipocytes, the other part being produced by other cells, particularly macrophages. 20 In vitro, IL-6 production by adipocytes is strongly increased by TNFα. 21 The respective role of TNFα and IL-6 produced by adipocytes and macrophages in WAT and during obesity-related inflammation, is difficult to estimate precisely. Nevertheless, as we will describe in section 2, both cells function in a coordinated manner, and macrophage recruitment in WAT is largely attributable to factors secreted by adipocytes, such as MCP-1. 22
The circulating levels of leptin and adiponectin, two hormones predominantly secreted by adipocytes, are respectively increased 23 and decreased 24 in obesity. Interestingly, these factors were shown to exert opposite effects on inflammation and on immune response. Leptin was initially described as an adipostat signal, secreted in proportion to adipose mass and controlling appetite and body weight in both humans and rodents. 25 The importance of leptin in immunity (reviewed in 26 ) was first revealed in obese mice with homozygous mutation in leptin (ob/ob) or leptin receptor (db/db), in which impaired immune responses were evidenced. 25 28 Our group, as well as others, has recently further clarified these immune dysfunctions by demonstrating that obese condition is associated with impaired functionality of T-lymphocytes, dendritic cells and macrophages, 29 31 cells of respectively, adaptive and innate immune systems. The demonstration of increased leptinemia during infection and inflammation further reinforced the role of leptin in inflammation and immunity. 32
Most if not all cells involved in innate immunity express leptin receptor at their surface and thereby are sensitive to leptin (reviewed in 25 ). Leptin induces activation of phagocytosis by monocytes/ macrophages and their production of nitric oxide and pro-inflammatory cytokines; induces the chemotaxis of neutrophils and the release of oxygen radicals; up-regulates natural killer (NK) cell-function including proliferation and cytotoxic activity by improving the expression of IL-2 and perforin; and stimulates the production of growth hormone by blood mononuclear cells, allowing survival and proliferation of immune cells. Leptin also modulates adaptive immune responses by interfering in the proliferation of certain sub-populations of T-cells: leptin increases the proliferation of naive CD4+ T-cells, whilst inhibiting the proliferation of memory CD4+ T-cells. In addition, leptin promotes T helper (Th)1- and suppresses Th2- type response (reviewed in 25 ).
In opposite to leptin, adiponectin exhibits potent immunosuppressive and anti-inflammatory properties. 6 Adiponectin impairs the production of TNFα, IL-6, IL-8 and interferon γ (IFNγ) by activated macrophages while inducing the production of anti-inflammatory mediators IL-10 and IL-1 receptor antagonist (IL-IRa) by monocytes/ macrophages and dendritic cells. The inhibition of nuclear factor kB (NF-kB) by adiponectin might explain some of these effects. 33 Interestingly, adiponectin modulates endothelial inflammatory responses by reducing the induction of adhesion molecules (ICAM-1, VCAM-1 and E-selectin), and therefore interferes with the adherence of monocytes to endothelial cells and their subsequent migration to sub-endothelial space. 34 This effect, added to its inhibitory capacity on transformation of macrophages to foam cells and on proliferation and migration of smooth muscle cells, has led to consider adiponectin as a potential anti-atherogenic factor. Adiponectin is the most abundant adipokine secreted by adipocytes and is present in high concentrations in the blood of healthy subjects (in the range of μg/ml versus ng/ml for leptin), thus weakening the impact of limited vascular injury. Although produced by adipocytes, its serum level significantly drops in obese patients, 24 and this was correlated with the development of cardiovascular disease observed in obesity.
In conclusion, adipose tissue might be regarded as an important in vivo source of inflammatory products (e.g. TNFα, IL-6, leptin and adiponectin) and could therefore actively participate to the initiation and the regulation of immune response and inflammation.
Moreover, adipocytes can be regarded as true innate immune cells: indeed, they expressed some receptors of innate immune system, which could enable them to both sense and respond specifically to any danger signals.
Receptors of the Innate Immune System are Expressed on Adipocytes: An Emphasis towards the Toll-Like Receptors
The innate immune system is the body's first line of defense against microbial, chemical and physical injury, whereby various reactions repair damage, avoid or isolate threats and restore homeostasis. In vertebrates, innate immunity is dependent in large part on myeloid cells that include mononuclear phagocytes, macrophages deriving from blood monocytes, and polymorphonuclear phagocytes. Sentinel trouble-shooting macrophages, as well as other immune cell-types, detect environmental threats through pattern-recognition receptors (PRRs) and release pro-inflammatory cytokines like IL-6 and TNFα.35,36
To date, the best characterized PRRs are Toll-like receptors (TLRs), a family of transmembrane receptors that is remarkably conserved from plants to vertebrates. 37 TLRs are broadly expressed in the cells of innate immune system such as macrophages and dendritic cells, but also in epithelial and endothelial cells and in organ parenchyma cells and TLRs have therefore specific roles in local innate immune defense. 38 Furthermore, the two major cell-types of adaptive immune response, i.e. T- or B-lymphocytes, express certain TLRs and respond directly to corresponding ligands in concert with triggering, respectively, T-cell and B-cell receptors. Thus, in addition to their well-described role in innate immunity, TLRs are also crucial in shaping adaptive immune response from its initiation to the development of immunological memory. 39
Interestingly, in addition to their role in innate and adaptive immunity, TLRs have recently been described to regulate bodily energy metabolism, mostly through acting on adipose tissue. Indeed, it was reported that TLR4 (sensing lipopolysaccharide (LPS) and saturated fatty acids) is expressed in the murine pre-adipose cell line 3T3-L1. 40 Interestingly, LPS-treated adipose cells secrete increased amounts of TNFα, and subsequently express higher levels of TLR2 (sensing bacterial lipoproteins). Recently, the presence of functional TLR2 and TLR4 was reported on human adipocytes isolated from subcutaneous fat tissue, 41 and several TLRs (TLR1 to 9) were found on mouse adipocytes.42,43 The activation of adipocytes via TLRs (mostly TLR4) results in the synthesis of pro-inflammatory factors such as TNFα or IL-6, and of chemokines such as MCP-1 (also known as CCL2), CCL5 or CCL11.40,41,44 Conversely, adipocyte-specific knockdown of TLR4 (e.g. shRNAi for TLR4 in 3T3-L1 cells; or adipocytes from TLR4-deficient mouse) prevented LPS-induced cytokine expression. Finally, adipocytes isolated from diet-induced obese mice or genetically obese animals (ob/ob or db/db mice) exhibited increased TLR expression,43,45,46 together with higher inflammatory cytokine production upon stimulation. 43 Of note, increased endotoxemia was observed in mice on high fat-feeding. Moreover, metabolic endotoxemia induced by a continuous LPS infusion had comparable effect on mouse body weight and glucose parameters (e.g. glycemia and insulinemia) to that of high-fat diet. 47 Mice genetically deficient in TLR4 or in CD14 (a co-receptor for TLR4) were reported to be of “ideal body type”: when fed with a chow diet, these mice exhibit increased bone mineral content, density and size, as well as decreased body fat. 48 Moreover, these mice do not become obese with age, unlike many strains of laboratory wild-type mice. This “perfect” phenotype of low adiposity and strong bones, with normal activity and fertility was baptized as the “Adonis phenotype” and this concept is currently further explored for its potential in the treatment of obesity.
However, this approach has to be considered with caution since contradictory results have been more recently obtained with high-fat-fed TLR4-deficient mice. Indeed, whilst some reports described no effect on body weight, 49 51 other authors described increased body weight 45 or, in contrast, protection against diet-induced obesity. 52 Similarly, adiposity and food intake were either reported to be unchanged, increased or decreased in TLR4-deficient animals.45,49–52 These divergent phenotypes could derive from the use of different mouse genetic backgrounds, different TLR4 mutation strategies or different feeding protocols (e.g. diet composition and timing). Despite these discrepancies in body weight and adiposity levels, they all revealed a marked improvement in insulin sensitivity when TLR4 gene was disrupted. Therefore TLR4, which is expressed in most tissues of the body, including the insulin sensitive ones such as adipose tissue, muscle and liver, 52 appears to be an essential mediator of bodily insulin-resistance.
TLRs are mostly expressed on innate immune cells such as macrophages and, as reported above, on pre-adipocytes and mature adipocytes. Interestingly, it should be mentioned that pre-adipocytes were shown to be able to convert into macrophage-like cells. 53 Indeed, adipocytes and macrophages share macrophage-specific antigens and the differentiation and function of both cell-types is controlled by PPARγ. 54 It has therefore been suggested that adipocytes and macrophages might be closely related and possibly interconvertible. Even still debated, this possible conversion between adipose cells and macrophages might nevertheless reinforce the view of adipose tissue as an integral part of innate immune system.
Taken together, the expression of functional TLRs on adipocytes classifies adipose tissue as a new member of innate immune system that is able to respond specifically to microbial or physical insults. This concept opens a new and fascinating perspective on a potential role of adipose tissue in host defense.
The second part of the review will show that adipose tissue is also an important site of inflammation and can recruit immune cells. Indeed, obesity and insulin-resistance have been closely associated to a massive infiltration of pro-inflammatory macrophages that initiates and sustains inflammation in obese adipose tissue.
Cells from Both Innate and Adaptive Immunity Initiate White Adipose Tissue Inflammation (Fig. 1)
Macrophages: Infiltration and Activated State in Obese Adipose Tissue
Distributed throughout the body, macrophages are a fundamental part of the immediate innate defense mechanisms, which can promote specific adaptive immunity by inducing T-cell recruitment and activation. Due to their collaboration with T- and B-cells, mostly through cell-to-cell contacts, macrophages also play an essential role in triggering, instructing and terminating the adaptive immune response.
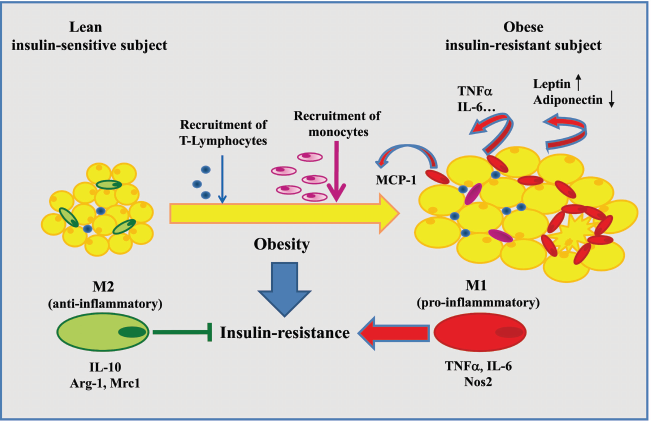
Adipokines and immune cells involved in obesity-induced adipose tissue inflammation and insulin-resistance development.
Depending on their tissue localization (lung, brain, liver or adipose tissue) and on the immunological micro-environment, macrophage activation can be either pro- or anti-inflammatory,55,56 thereby supporting the activation of the respective T helper (Th) cell-subsets Th1 or Th2, as defined by Mosmann et al. 57
In adipose tissue, macrophages were shown to play a key role in the development of the inflammatory state associated with obesity. Indeed, macrophages accumulate in the adipose tissues of various obese mouse models (such as the diet-induced obesity or the genetically deficient model in the leptin gene ob/ob).9,58 The number of macrophages positively correlates with body mass and adipocyte size in both subcutaneous and visceral fat depots, even though macrophage infiltration is more prominent in the latter. Similar relationships were confirmed in human subcutaneous 9 and visceral 59 adipose tissues. Accumulated macrophages are considered to be the critical link between obesity and inflammation since they are the major source of pro-inflammatory cytokine production, notably TNFα and IL-6, in adipose tissues.9,58,60 Interestingly, the process appears reversible since macrophage infiltration and pro-inflammatory marker expression in the adipose tissue of obese subjects can be significantly reduced after weight loss.9,58,60,61 Similarly to any immune and inflammatory response, macrophage infiltration in expanding adipose tissue results from blood monocytes influx, likely attracted by the chemokine MCP-1.9,62 Indeed, it was reported that MCP-1 secretion is markedly enhanced locally and in plasma in obese rodents58,22 and human patients. Over-expression, deficiency or mutation-induced dysfunction of MCP-1 in different mouse models interfere with adipose tissue macrophage (ATM) accumulation along with insulin-resistance development. 22 While a pivotal role for MCP-1 and its receptor CCR2 is strongly suggested, 63 recent studies have challenged this view by reporting no noticeable impact for the genetic disruption of the chemokine regarding macrophage accumulation and glucose tolerance improvement.64,65 Among various potential factors, other chemoattractant cytokines such as osteopontin 66 and granulocyte-macrophage colony-stimulating factor (GM-CSF) 67 could also participate in macrophage infiltration during high-fat diet-induced obesity. MCP-1 and osteopontin are rather produced by the various cell types composing the stromal vascular fraction of adipose tissue than by mature adipocytes,58,60,68,66 questioning the causal relationship between obesity-associated adipocyte perturbations and macrophage recruitment. Indeed, adipose tissue expansion is not systematically associated with macrophage infiltration and insulin sensitivity impairment. In a mouse model of morbid obesity (consecutive to adiponectin over-secretion in plasma), a massive development of adipose tissue corresponding to adipocyte hyperplasia did not induce macrophage accumulation. The authors suggested that the integrity of adipocyte function may be a more critical determinant of the local inflammation than the increased adipose mass per se. 69 The precise nature of the event priming adipose tissue inflammation definitively remains to be determined even though some hypotheses have already been proposed. For example, it was suggested that leptin may activate endothelial cells to facilitate monocyte diapedesis. 62 A process of necrosislike cell death of adipocytes could also induce macrophages recruitment to phagocyte cell debris. Indeed, several immunohistochemical analyses have consistently reported ATM organization into “crownlike” structure around dead adipocytes in adipose tissue of obese rodents70,71 and humans. 60
More recently, some mechanistic insights to explain how macrophages are associated with inflammation in obesity, have emerged from the characterization of ATM activation state. Macrophage population and function have been revealed to be highly heterogeneous and dependent on the surrounding environment, which has led to their characterization and classification following the well-known classification of T-cell activation state into Th1/Th2 sub-types. 72 Many refer to polarized macrophages as M1 and M2 cells with distinct functions that are elicitated in response to the factors that dominate the inflammatory scene. Typically, macrophages can be distinguished between the M1 phenotype, identified as the proinflammatory or “classically”-activated state, secreting various cytokines (e.g. TNFα, IL-6), and the M2 phenotype referred as to the anti-inflammatory or “alternatively”-activated state, which produces IL-10 and TGFβ. Cytokines such as IFNγ, secreted by Th1 cells, or IL-4 and IL-13, produced by Th2 cells control the M1/M2 polarity, respectively favouring a classical activation or an alternative activation of macrophages. M1 and M2 activation states are mainly characterized according to distinct gene expression patterns reflecting their inflammatory activities (e.g. inducible nitric oxide synthase Nos2, cyclooxygenase Cox2) or their tissue remodelling and reparation properties (e.g. arginase-1, mannose receptor).
Following a pulse dye labelling of ATM to discriminate newly infiltrated ATM from the resident ATM, it has been shown that recruited ATM during a diet-induced obesity exhibit an inflammatory M1 profile compared to the already settled ATM. 73 Moreover, the same group has recently demonstrated that ATM from lean mice retain a gene expression pattern typical of the M2 activated state, while ATM from obese mice are characterized by enhanced expression levels of TNFα and Nos2, both markers of the M1 activated state. This study supports that diet-induced obesity either converts or promotes the replacement of initial M2-polarized ATM by M1-polarized ATM, thereby contributing to the development of insulin-resistance. 74 Interestingly, fully differentiated 3T3-L1 adipocytes and mouse primary adipocytes were recently shown to release significant amounts of IL-13 and, to a lesser extent, to produce IL-4 75 suggesting that adipocytes themselves could maintain ATM in the M2 polarity. Furthermore, the M1/M2 balance and intrinsic metabolism of macrophages appear closely intertwined. Indeed, the M2 state programming seems highly dependent on a metabolic switch of macrophages toward fatty acid oxidation: upon IL-4 exposure, bone-marrow-derived macrophages undergo gene expression changes favouring (3 oxidation and mitochondrial biogenesis; in contrast, oxidative metabolism blockade prevents the IL-4-mediated switch toward an anti-inflammatory phenotype. 76 A potential relationship between metabolism and macrophage differentiation state has recently been strengthened through the assessment of the role of two isoforms of PPAR: PPAR γ and δ/β are nuclear transcription factors controlling the expression of genes involved in adipogenesis and fatty acid oxidation/oxidative metabolism. 77 79 Both PPARγ and PPARδ/β are expressed in macrophages where their expression can be respectively induced by IL-4 76 and IL-13. 75 Macrophage-specific disruption of the PPARγ gene 80 or the PPARδ/β gene 75 severely alters the gene expression signature corresponding to the M2 state, promotes M1 polarization and concomitantly worsens insulin sensitivity impairment in mice. The relative contribution of each PPAR isoform as well as the nature of the factors (Th2 cytokines versus lipid ligands) triggering PPAR activation in macrophages remain to be further established. However, these studies emphasize that metabolic pathways and their regulating transcriptional factors can modulate the immune functions of macrophages.
In conclusion, ATM are the major source of pro-inflammatory mediators in obese adipose tissue and contribute both to the local and systemic metabolic alterations and to the general inflammatory state. Their number and activation state are likely crucial in the onset of obese tissue inflammation and in the development of insulin-resistance. Nevertheless, it should be stated that most of these recent findings are based on ATM phenotyping in several murine models and their relevance to human obesity and its metabolic complications have not yet been fully addressed. Indeed, ATM from obese patients and mice release the same pro- (TNFα, IL-6, MCP-1) and anti- (IL-10, TGFβ) inflammatory factors58,74,60,81 but differ with respect to other M1/M2-associated markers such as Nos2 and arginase-1. 81 Key questions are therefore still remaining unanswered such as: What are the mechanisms triggering macrophage intervention and governing the “selection” between M1 or M2 phenotype? Is there coordination between adipocytes and macrophages to regulate their metabolic and immune/inflammatory responses in the context of obesity and its related complications?
Recent studies have shown that, besides their classification as M1 or M2-orientated cells (cf
Lymphocytes Might Precede Macrophage-Infiltration in Obese Adipose Tissue
Whereas T-cells are undoubtedly involved in the regulation of inflammation in atherosclerosis, their role in adipose tissue inflammatory process has just begun to be investigated. Immunohistological and flow cytometry analyses have recently revealed the presence of resident lymphocytes (identified as CD3+-T-cells and CD19+-B-cells) in mouse visceral and subcutaneous adipose tissues. 84 Even though first studies failed to detect any change in T-cell number in adipose tissue of obese mice,58,84 several recent studies have pointed out that dietary 85 87 or genetic 112 obesity is also associated with T-cell infiltration including both CD4+ and CD8+ cells. Immune cell composition assessment at an early stage of high-fat diet-induced obesity suggests that T-cell entry in adipose tissue could precede monocyte attraction and, therefore, might represent one of the processes initiating adipose tissue inflammation. 86 Indeed, both the secretion of the chemokine CCL5/“Regulated on Activation, Normal T-cell Expressed and Secreted” (RANTES) and the expression level of its receptor CCR5 are enhanced in adipose tissue of obese male mice. 85 Moreover, RANTES neutralization reduces, in vitro, T-cell migration. 85 Interestingly, RANTES expression was not only restricted to T-cells but was also detected in mature adipocytes, more prominently in presence of TNFα. 85 If the mechanisms underlying T-cell attraction in adipose tissue are far to be deciphered, even less is known regarding their activation state in the obesity-associated inflammation. However, the implication of the Th1-type cytokine IFNγ in adipose tissue inflammation has recently been explored. 87 IFNγ mRNA expression was up-regulated in mouse adipose tissue after high-fat diet feeding. 3T3-L1 adipocytes incubated with IFNγ exhibited a marked enhancement of the expression level of various cytokines and chemokines. Moreover, IFNγ deficiency partly prevented the diet-induced increase of both ATM number and pro-inflammatory marker expression (TNFα and MCP-1) in adipose tissue. This phenotype was associated with a slight improvement of glucose intolerance. Overall, this first characterization supports that IFNγ could participate in obesity-associated inflammation. 87 Eventually, the involvement of other factors known to drive Th1 activation and locally produced in WAT, such as leptin, 88 would need to be questioned regarding the activation of lymphocytes within the fat tissue.
Hormonal Modulation of White Adipose Tissue Inflammation: A Focus on Glucocorticoids
Cortisone is a glucocorticoid (GC) hormone that was first used to treat rheumatoid arthritis in humans in the late 1940s, thereby leading to the discovery of the anti-inflammatory effects of GC. Endogenous GC, however, are rather immunomodulatory than simply anti-inflammatory. Depending upon concentration and timing, GC either enhance or suppress immune responses, thus shaping both innate and adaptive immunity. Strikingly, prolonged exposure to GC, as seen in Cushing's syndrome, leads to morphological and metabolic features resembling those of the metabolic syndrome. 89 Indeed, Cushing patients develop upper body obesity, hypertension and hyperglycemia. Excellent reviews dealing with the effects of GC on metabolism have recently been published. 90 93 Therefore, considering the scope of our review, we will mainly focus on the modulation of inflammation by GC, likely participating in local inflammation within obese adipose tissue.
During inflammation, GC were reported to promote differentiation and survival of anti-inflammatory macrophages (M2; cf
Importantly, the link between 11β-HSD1 expression and fat mass has also been reported in human obesity. Both subcutaneous and visceral adipose tissues of obese individuals were shown to express higher levels of 11β-HSD1 than those of lean subjects.98,99 It has been proposed that adipose tissue-specific rise in 11β-HSD1 in obesity might amplify intracellular glucocorticoid levels, consequently modulating the transcription of several key target genes involved in adipocyte and macrophage functions.
Modulation of WAT inflammation by glucocorticoids is likely not exclusively restricted to an effect on adipocytes. GC could also affect immune cells that are present in adipose tissue, especially macrophages that are essential in WAT inflammatory process (cf
Altogether, these recent evidences pointing out the modulatory role of 11β-HSD1 on inflammation, suggest that the inhibition of 11β-HSD1, specifically in adipose tissue, could represent a novel therapeutic strategy to treat metabolic diseases such as obesity.
Conclusive Remarks
Obesity is a world-wide epidemic currently viewed as a state of chronic, low-grade inflammation characterized by a pro-inflammatory alteration in the serum cytokine profile as well as an infiltration of white adipose tissue by activated macrophages.9,59 A better knowledge of how inflammatory pathways are chronically activated is crucial since inflammation undoubtedly contributes to insulin-resistance and type-2 diabetes. Adipose tissue inflammation could exacerbate (notably through TNFα release) adipocyte metabolic and endocrine dysfunctions, both participating to the development of insulin-resistance and type 2 diabetes. Indeed, the impairment of free fatty acid storage in adipose tissue can also be involved in the loss of peripheral insulin sensitivity and insulin secretion: reduced fatty acid uptake and increased lipolysis can lead to inappropriate accumulation of lipids in non-adipose tissues such as liver, skeletal muscles and pancreatic islets, subsequently leading to defects in insulin action and secretion (reviewed in102,103).
Leptin and adiponectin participate to the control of glucose homeostasis104,105 and the secretion level of both adipokines is altered in obesity.23,24 Interestingly, the alterations of adipose tissue metabolic and endocrine functions likely participate to the development of insulin-resistance and type 2 diabetes that occurs in lipodystrophic patients who are characterized by a selective loss of adipose tissue (reviewed in106,107). Due to its low incidence and high heterogeneity, the lipodystrophic syndrome has not been as well-explored as obesity and studies regarding the inflammatory status of the remaining fat depots of lipodystrophic patients are currently missing.
It has also to be emphasized that obesity, defined by a high body mass index and an elevated fat mass, is not systematically associated with insulin-resistance, metabolic or cardiovascular complications. As consistently shown, accumulation of visceral fat is more detrimental than accumulation of subcutaneous fat. Moreover, the characterization of different subtypes of obesity has recently led to the identification of a subset of obese, yet highly insulin-sensitive, patients (reviewed in 108 ). These so-called “metabolically healthy but obese” individuals display less visceral fat mass 109 and lower plasma levels of inflammatory markers (e.g. IL-6 110 and C-reactive protein 111 ) than unhealthy obese individuals. Further work on the inflammatory state (e.g. macrophage number and activation state, cytokine production) of the adipose tissue of the “metabolically healthy but obese” patients is needed to clearly define whether this sub-population of obese individuals is also protected from adipose tissue inflammation. However, obvious ethical reasons and difficulties to diagnose and “classify” lipodystrophic and obese patients are real limits to such investigations. This partly accounts for the common use of animal models, which have their own limitations due to inherent species- and/or strain-based differences and to difficulties to entirely reproduce the etiology and pathological profiles associated to human adipose tissue disorders.
Beside the hormonal regulation of adipose tissue mass and inflammation (that we exemplified by the exciting results on glucocorticoids), the inciting event that triggers the inflammatory cascade in adipose tissue remains to be elucidated. Yet we reported that a current hypothesis for the accumulation of macrophages within obese white adipose tissue might be the recruitment of T-lymphocytes85–87,112and possibly of dendritic-like cells,74,73,82,81 prior to that of macrophages. Nevertheless macrophages—which accumulate within the adipose tissue of both obese rodents and humans, switch their functional phenotype from M2 to M1,74,73 and produce several pro-inflammatory molecules-are believed to significantly modulate this process.
Besides, we also reported that adipocytes express innate receptors such as the Toll-like receptors, mostly TLR4. 38 TLR4 is the receptor for bacterial LPS and a key molecular component of innate immune system which function was most expensively studied in macrophages. 113 We described how TLR4 also contributes to insulin-resistance 52 and how TLR4 stimulation activates pro-inflammatory pathways similar to that encountered in obese tissues. 114
Altogether, we attempted in this review to show that adipose tissue is part of innate and adaptive immune systems. Indeed, it produces inflammatory cytokines (TNFα, IL-6), as well as factors regulating monocyte/macrophage function (leptin, adiponectin). Inflamed adipose tissue is massively infiltrated by cells of both innate and adaptive immune system such as macrophages and possibly T-cells and dendritic-like cells. The expression of a broad spectrum of innate immune receptors such as Toll-like receptors on both pre-adipocytes and mature adipocytes, together with a possible conversion of the former into macrophage-like cells, 53 collectively establish white adipose tissue as a new member of the immune system and might position this tissue as being “the key road to inflammation”. Adipose tissue is well-spread in the body and could provide immune cells and/or factors which could participate to local inflammation or immune response, as recently proposed for Crohn's disease. 115 Broadly, it opens new and fascinating perspectives on a potential role of adipose tissue in host defense and inflammatory disease.
Disclosure
The authors report no conflicts of interest.
Footnotes
Acknowledgments
The authors were supported by the Centre National de la Recherche Scientifique (CNRS; to IW), the Institut Pasteur of Lille (to CV) and the Région Nord-Pas de Calais (to SL). We also thank Karl Oulmi (IFR 142) for the Artwork.