
Abstract
This study was conducted with the aim of optimizing the experimental design of array experiments. We compared two image analysis and normalization procedures prior to data analysis using two experimental designs. For this, RNA samples from Charolais steers
Introduction
Microarray technology is gaining ground as an approach for exploring major subsets or almost complete gene profiles of organisms. The technology makes it possible to analyze a variety of conditions such as samples of several treatments, mutants, developmental stages or time points. However, gene expression values derived from microarrays are often considered less reliable than other biochemical data (Mangalathu et al. 2001). Experimental biases in gene expression profiling could occur due to varying total amounts of hybridized mRNA, different label incorporation rates, the methodology applied for spot quantification trough image analysis, or bleaching effects of the dye. The comparability and reliability of data generated using microarray technology could be enhanced by using of a common set of standards, as recently proposed (Shi et al. 2006), which would allow better accuracy and reproducibility as well as dynamic range assessments, although probably not without certain persistent problems.
Variability in microarray experiments can arise from either pre-scanning steps (methods of RNA extraction, different types of probe preparation, probe labeling, hybridization and slide quality) and/or post-scanning steps (image acquisition and image/data analysis), as previously reported (Ahmed et al. 2004). Relatively little attention has been given to the variability introduced by image analysis methods as a potential source of noise. Variability introduced by image analysis may predominantly be generated by the method used to estimate signal background from a spot and segmentation. Another important point to emphasize is that the one-color and two-color designs yield equivalent data, yet this variable is not considered as a critical factor (Patterson et al. 2006).
Normalization methods have been developed for assessing spot quality in an extended examination factoring in spot size, signal-to-noise ratios, background uniformity, and saturation status. For segmentation methods, examples of algorithm and software implementations have been described elsewhere (Yang et al. 2001), and comparisons have been performed between the algorithms used by different segmentation methods (Yang et al. 2000). It has been shown that small and large-scale fluctuations in pixel intensities within a spot lead to uncertainty in microarray quantitation, and that pixel-to-pixel variability correlates with variability between replicate spots on duplicate slides (Brown et al. 2001). To underline these issues, most investigators use the commercial software provided (for instance
The aim of the statistical tests (Student's
The aim of the research reported here was to assess different microarray procedure, based on different image analysis and statistic methods by focusing on variability in the results. We have analyzed two different datasets (obtained after hybridization of the same samples on the same slides with two experimental designs) using two image analysis/normalization methods and two statistical approaches. We show that using software such as
Materials and Methods
Biological samples
For the present study, we used
RNA isolation and labeling
Total RNA was extracted from SC adipose tissue as described previously (Bonnet et al. 1998) and extracted from LT muscle using TRIzol Reagent (Life Technologies, UK). The total RNA was then purified and treated with DNAseI using the RNeasy® Mini kit (Qiagen, France) as recommended by the manufacturer. RNA integrity was checked using Lab Chip Agilent technology, as previously described (Bernard et al. 2007). The reference corresponded to a mixture of same quantities of total RNA from SC, LT and mammary gland samples. Mammary gland RNA was used as a positive control since 6 genes are highly expressed in this organ.
Microarray hybridization and data acquisition
Labeling was performed using a “Pronto Plus Direct Systems” kit (Corning-Promega Europe, France) according to the supplier's guidelines. Five µg of total RNA was reverse transcribed into cDNA in the presence of cyanine (Cy3-dCTP/Cy5-dCTP, Amersham, UK). cDNA purification was performed according to the manufacturer's instructions, and concentration and frequency of incorporation were determined by spectrophotometry. Cyanine 3 and cyanine 5-labelled cDNA (40 pmol each) were mixed, dried and resuspended in 40 µl “long-oligo” buffer before hybridization onto one microarray (8400 probes from Operon, Corning Ultra GAP-spotted in Genopole Toulouse, France). Hybridization was performed at 42 °C for 16 h as previously described (Ollier et al. 2007). Post-hybridization washes followed by slide centrifugation were carried out before the scan. The experimental design (Fig. 1) was as follows for each sample (hybridization 3 from triplicate was used as dye swap in the data analysis). Hybridized microarrays were scanned with a 428 MWG Array Scanner (MWG Biotechnologies).
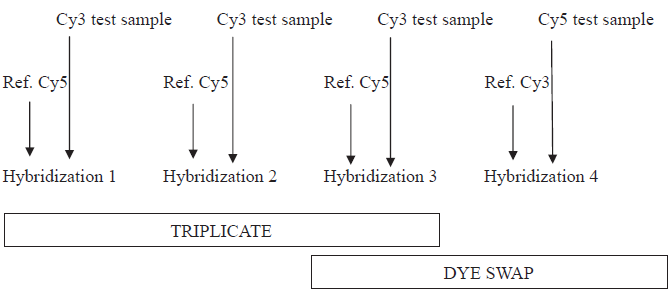
Experimental design used in this study. Test sample is RNA of the target sample (LT and SC) to be studied and Ref. is the reference (RNA from LT, SC and mammary gland).
Data analysis
Image analysis was achieved using two different software solutions:
Raw data were filtered and normalized using either
Hybridization data were run through SAM (Significance Analysis of Microarray) analysis (Tusher et al. 2001) or multi-test model. The last statistical analysis was performed after filtering using R 2.1 software: after controlling the variance of each gene, log of ratios between Sample and Reference were analyzed with an ANOVA model by using standard Student's
Control and common genes were determined between the results from
Results
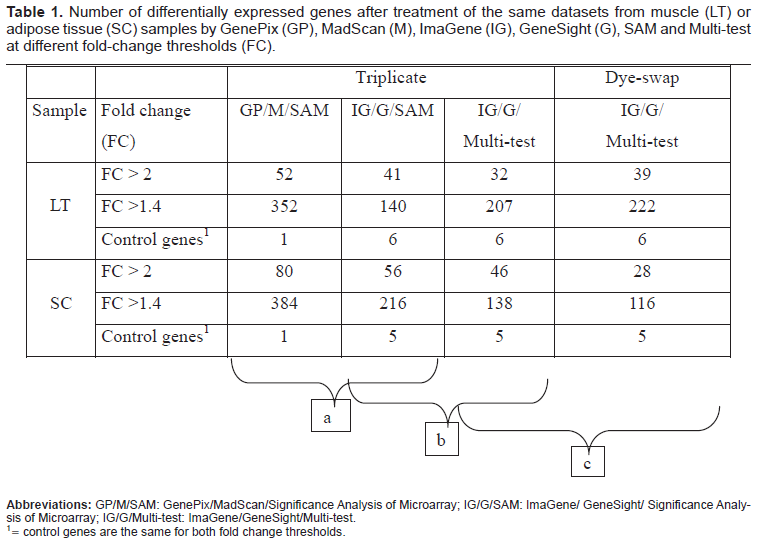
The number of differentially expressed genes with each procedure is summarized in Table 1. Globally for high fold change (FC > 2), both the combination of image analyses and normalization using
Number of differentially expressed genes after treatment of the same datasets from muscle (LT) or adipose tissue (SC) samples by GenePix (GP), MadScan (M), ImaGene (IG), GeneSight (G), SAM and Multi-test at different fold-change thresholds (FC).
To have positive control, we have implemented the reference sample with mammary RNA. Milk proteins genes (CSN1S1, CSN1S2, CSN2, CSN3, LALBA and LGB) were specially expressed in the mammary gland of lactating cow. Thus, compared to LT or SC tissues, these genes could be used as positive control of differentially expressed genes. We observed that the differential expression on these 6 genes depended on the combination of the image analysis system with the normalization procedure used.
Within the triplicate experimental design, only one gene (CSN2, FC = 5.4) was declared differentially expressed with the
Comparison of two image analysis/normalization procedures
Under the triplicate design and with the same statistical procedure (SAM), the number of genes that exhibited statistically significant changes (p < 0.001) in expression (FC > 1.4) was 352 for the
Comparison of two statistical methods
Statistical analysis was evaluated under the triplicate experimental design, after image analysis and normalization procedures by
Comparison of two experimental designs
Finally, we have compared triplicate and dye-swap experimental design using the same process of data
Discussion
The purpose of this study was to compare two post-scanning methods
Importance of image analysis and data normalization
The comparison of two post-scanning methods prior to statistical analysis (
The combination of image analysis and data normalization is indeed an important aspect of microarray experiment. These steps can have a potentially large impact on downstream analyses such as the identification of differentially expressed genes. A number of microarray image analysis packages (commercial software and freeware) are now available. GenePix works along a principle based on fixed-circle and adaptive circle methods whereas
Importance of statistics
Designing procedures to control the FDR (criterion for identifying differentially expressed genes) are challenging problems (Verducci et al. 2006). SAM and multiple t-tests use R and its Bioconductor package. Before SAM can be run, there are some permutations that are needed to generate FDR. The Bonferroni, is a Family-Wise type Error Rate (FWER) method to correct for multiple testing to eliminate false positives (Lin, 2005). We aimed to compare the widely used SAM and the multi-test with p-value adjusted using Bonferroni correction, since there is no reference method to control the false positives. Based on our results, there was no significant statistical treatment-related difference in the number of differential genes.
Importance of the experimental design
First, for the two samples studied, we proposed a “reference design” including an experimental procedure based on replicate spots within each microarray. We compared hybridization to a reference sample with either a triplicate or a dye-swap design, as shown in Figure 1. This procedure allows us to minimize biological noise by reducing individual variability through testing sample-specific dye bias. We did not identify any significant difference between triplicate and dye swap, which is consistent with a previous study (Dobbin et al. 2005) reporting that sample-specific dye biases appeared to have only minimal impact on estimated gene-expression differences. However, we found that dye swap sorted some genes than triplicate design, although from limited samples, thus showing a gain in cost with no loss in efficiency (Dobbin et al. 2003).
Conclusion
There are many sources of variability that can affect gene expression intensity measurements, but not all are well characterized or clearly identified.
With these results, we demonstrated the impact of conducting the data extraction step before applying statistical models. From the above discussion, it is obvious why there can be a great deal of discordance in results obtained equally as often from different microarrays as from within the same microarray platforms. Ultimately, the decision between triplicate or dye-swap, software image analysis and normalization approaches is more often driven by cost, experimental design considerations, and objectives of the study. This raises the question of how array data can be compared. By presenting the experimental design and performance advantages of both models, we have provided insight and guidance for properly selecting the best approach depending of the objectives of the study.
Footnotes
Abbreviations
Acknowledgments
This study was funded by a national grant from: the “Agence Nationale de la Recherche”and APIS-GENE (GENANIMAL call, national program AGENAE) for the project MUGENE (GENEs of the MUscle tissue). Ali Kpatcha Kadanga is funded by an INRA grant from the “Physiologie Animale et Système d'Elevage (PHASE)” Department.