With the advent of high throughput sequencing platforms and relevant analytical tools, the rate of microbial genome sequencing has accelerated which has in turn led to better understanding of microbial molecular biology and genetics. The complete genome sequences of important industrial organisms provide opportunities for human health, industry, and the environment. Bacillus species are the dominant workhorses in industrial fermentations. Today, genome sequences of several Bacillus species are available, and comparative genomics of this genus helps in understanding their physiology, biochemistry, and genetics. The genomes of these bacterial species are the sources of many industrially important enzymes and antibiotics and, therefore, provide an opportunity to tailor enzymes with desired properties to suit a wide range of applications. A comparative account of strengths and weaknesses of the different sequencing platforms are also highlighted in the review.
Whole genome sequencing is a powerful approach for gaining the reference sequence information of multiple organisms. It helps in identifying genomic variations associated with phenotypes. Whole genome sequencing enables the identification of individual nodes of multi-component genetic interaction networks simultaneously, and also helps in mapping the evolutionary pathways that can support the growth of a genetically compromised strain.
Whole genome sequences of industrially important organisms and bacterial pathogens provide prospectives for industry, human health, and the environment. A wide diversity is present in bacterial genomes. Most of the bacterial proteins are highly or moderately conserved in evolution as extensive gene shuffling occurs in a few conserved gene arrays in distantly related bacteria. The outcome of whole genome sequencing has revealed new insights into the evolution of bacterial lifestyles including strategies for adaptation to new niches and overcoming competitors.
Bacillus is the most important workhorse among industrial microorganisms. They have ancient history of more than thousand years since the production of natto (a traditional Japanese dish of fermented soya beans) using B. subtilis, and their roles are continuously expanding and evolving. Bacillus species are considered as generally regarded as safe (GRAS) in the list of Food and Drug Administration (FDA). They are known to secrete copious amounts of extracellular enzymes and have high growth rates and short fermentation cycles and therefore these are one of the most important industrial enzyme producers.1–5 For further development and exploitation of these bacteria in industrial processes, the complete genomes of several species of Bacillus have been sequenced in order to understand their biochemistry, physiology, and genetics. B. subtilis is the first gram positive bacterium whose genome was sequenced and since then, the genomes of several Bacillus species have been sequenced.
Strategies to Sequence Complete Bacterial Genomes
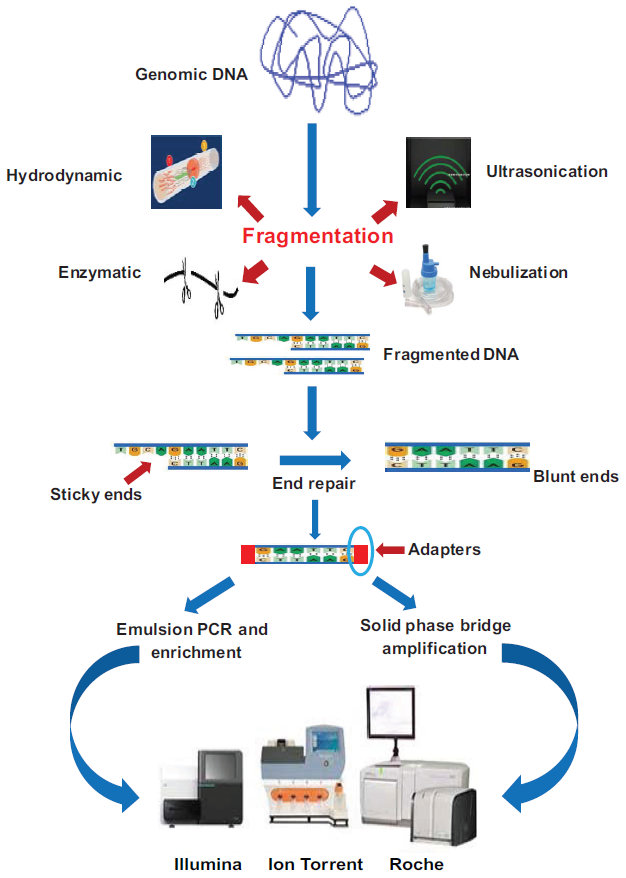
The random shotgun sequencing method was developed and perfected for prokaryotic genomes which are smaller in size and contain less repetitive DNA. Various methods are used to fragment DNA including nebulization, ultrasonication, enzymatic fragmentation, and hydrodynamic shearing. After fragmentation, adaptors are ligated to these fragments. Template amplification is the next step and most of the platforms support mate pair sequencing, in which the ends of DNA fragments of size 3–20 kb are connected to form circular molecules. These molecules are then again fragmented, and the fragments flanking the joints are then selected and the end adaptors are added. Such sequencing offers significant information about the location of sequences scattered across the genome, which aids in assembling the genome.6 Another platform is paired end sequencing which is similar to mate pair sequencing however in the former DNA fragments are sequenced from each end with no requirement for additional library preparation steps. The pictorial representation of the whole genome sequencing is summarized in Figure 1.
Pictorial representation of whole genome sequencing.
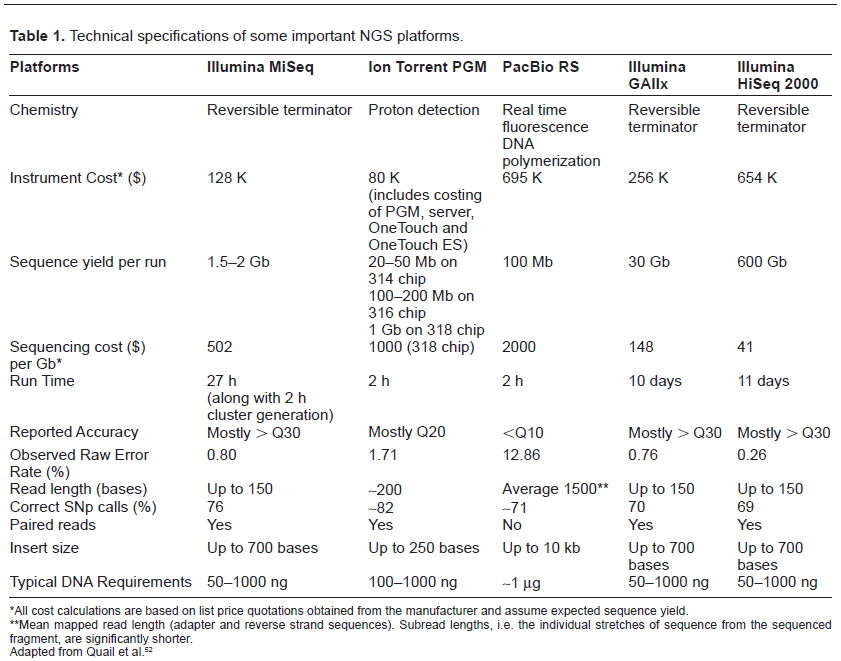
In 1977, Frederick Sanger introduced DNA sequencing technology, based on chain-termination method (also known as Sanger sequencing). Walter Gilbert developed another sequencing technology which relies on chemical modification of DNA and successive cleavage at specific bases. Sanger sequencing was accepted as the primary technology in the ‘first generation’ of laboratory and commercial sequencing applications on the basis of its high efficiency and low radioactivity. During that time, DNA sequencing was tedious and radioactive materials were required. The first automatic sequencing machine (AB370) was launched by Applied Biosystems in 1987, with capillary electrophoresis making the sequencing faster and more accurate. With the advent of new informatic approaches, holistic integration, and significant analysis of the genome sequence data, the sequencing has become easy and large information can be extracted from the complete genome. A variety of sequencing methods, softwares for the assembly of contigs, and annotation of gene are employed for sequencing the genome. The next generation sequencing (NGS) reads millions of fragments in a single run. It offers several advantages such as extensive parallel sequencing of shotgun libraries, high throughput with maximum genome coverage, and cost effectiveness. The most popular NGS platform includes (Roche 454: GS FLX, XL+; Illumina: Genome analyzer, HiSeq2000; Life Technologies: Ion Torrent, SOLiD, Pacific Biosciences: Pac Bio RS) sequencing. Illumina and Roche are the most commonly used platforms for sequencing genomes of Bacillus spp. The comparison of various sequencing platforms is depicted in Table 1.
Technical specifications of some important NGS platforms.
All cost calculations are based on list price quotations obtained from the manufacturer and assume expected sequence yield.
Mean mapped read length (adapter and reverse strand sequences). Subread lengths, i.e. the individual stretches of sequence from the sequenced fragment, are significantly shorter.
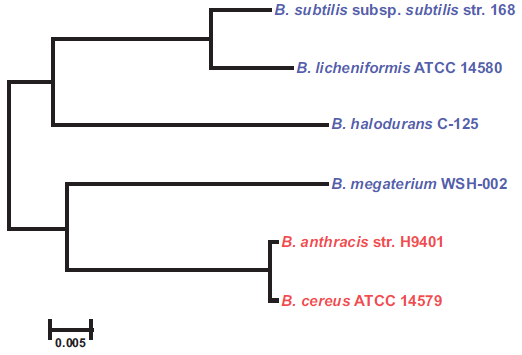
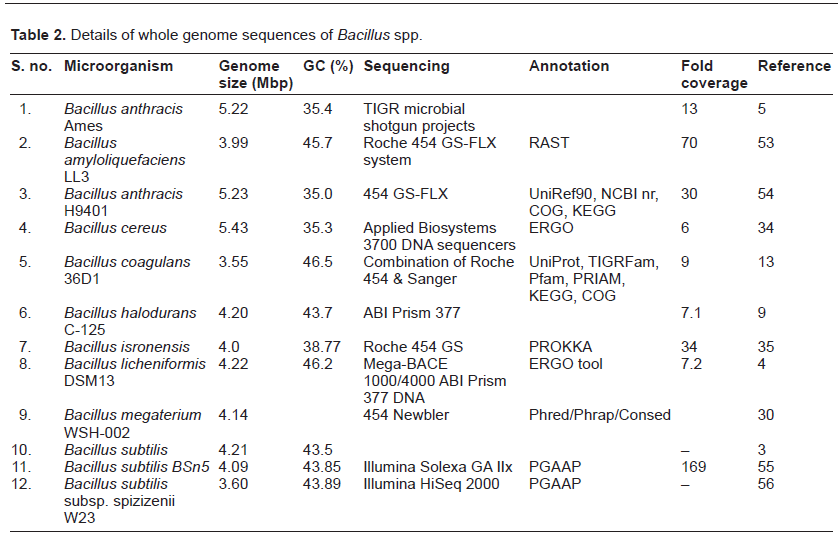
The first bacterial genome project started in 1991 for sequencing the complete Escherichia coli genome.7 Since then, genome projects have been started on a number of bacteria, archaea, and eukaryotes. Bacillus is one of the best described genera in the genomic database. According to Alcaraz et al8 more than 108 complete and draft genome sequences of Bacillus are available. The extensive research on Bacilli includes classical microbiology, biochemistry, and modern genomic and proteomic approaches. The details of genomes of various Bacillus spp. and their comparative analyses are highlighted in Tables 2 and 3. The dendrogram drawn using 16S rDNA sequences suggests close phylogenetic relationship among industrially important (B. licheniformis, B. subtilis, B. halodurans, B. megaterium) and pathogenic (B. anthracis, B. cereus) Bacillus spp. (Fig. 2).
Phylogenetic relationships among Bacillus spp. Closely related industrially important Bacillus spp. are highlighted in blue and the pathogenic Bacillus spp. are highlighted in red.
Details of whole genome sequences of Bacillus spp.
S. no.
Microorganism
Genome size (Mbp)
GC (%)
Sequencing
Annotation
Fold coverage
Reference
1.
Bacillus anthracis Ames
5.22
35.4
TIGR microbial shotgun projects
13
5
2.
Bacillus amyloliquefaciens LL3
3.99
45.7
Roche 454 GS-FLX system
RAST
70
53
3.
Bacillus anthracis H9401
5.23
35.0
454 GS-FLX
UniRef90, NCBI nr, COG, KEGG
30
54
4.
Bacillus cereus
5.43
35.3
Applied Biosystems 3700 DNA sequencers
ERGO
6
34
5.
Bacillus coagulans 36D1
3.55
46.5
Combination of Roche 454 & Sanger
UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG
9
13
6.
Bacillus halodurans C-125
4.20
43.7
ABI Prism 377
7.1
9
7.
Bacillus isronensis
4.0
38.77
Roche 454 GS
PROKKA
34
35
8.
Bacillus licheniformis DSM13
4.22
46.2
Mega-BACE 1000/4000 ABI Prism 377 DNA
ERGO tool
7.2
4
9.
Bacillus megaterium WSH-002
4.14
454 Newbler
Phred/Phrap/Consed
30
10.
Bacillus subtilis
4.21
43.5
–
3
11.
Bacillus subtilis BSn5
4.09
43.85
Illumina Solexa GA IIx
PGAAP
169
55
12.
Bacillus subtilis subsp. spizizenii W23
3.60
43.89
Illumina HiSeq 2000
PGAAP
–
56
Comparative analysis of some industrially important Bacillus spp.
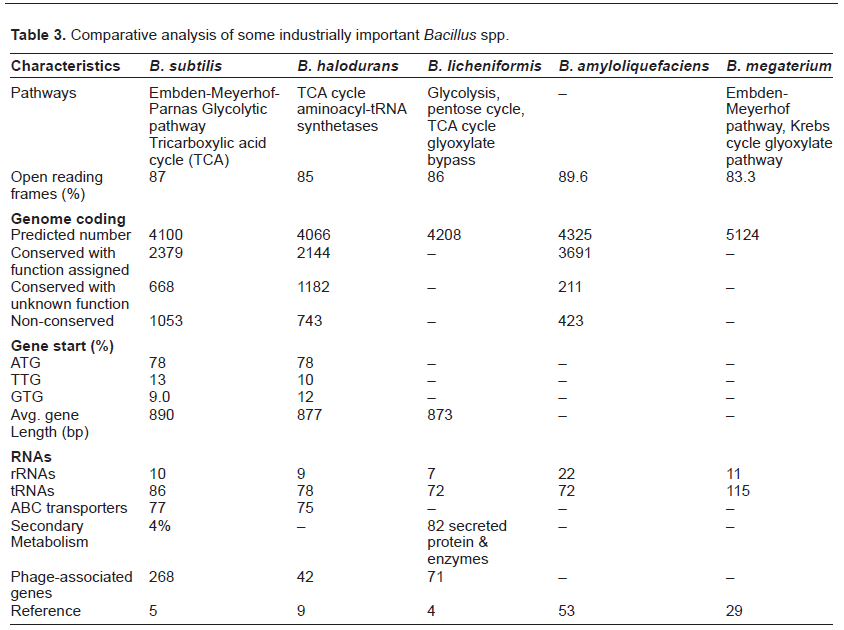
The type and range of metabolism provides information regarding the natural environment of the organism and biological activity. The key feature of B. subtilis metabolism includes the requirement of branched short chain carboxylic acids for lipid biosynthesis. It can also synthesize and utilize branched short chain carboxylic acids and alcohols.10 The high growth yield of B. licheniformis is due to its ability to utilize carbohydrates under conditions of varying oxygen tensions.11B. anthracis shows reduced ability for sugar utilization as compared to B. subtilis. It lacks catabolic pathways for mannose, arabinose and rhamnose, less phophotransferase system, and sugar transporters. However, the existence of genes for extracellular chitin and chitosan hydrolysis and N-acetylglucosamine utilization refects the association of this bacterium with insects.12B. cereus has fewer genes for the degradation of carbohydrates and therefore this group lacks the metabolic versatility for the uptake and assimilation of plant-derived carbohydrates present in the soil as compared to B. subtilis.13
Horizontal Gene Transfer
The comparisons among bacterial, archaeal, and eukaryotic genomes show that an important fraction of genes in the prokaryotic genomes have been subjected to horizontal gene transfer (HGT). The occurrence of HGT can be demonstrated by the discovery of pathogenicity islands, i.e., gene clusters that possess pathogenicity determinants in parasitic bacteria, and similar ‘symbiosis islands’ in symbiotic bacteria.14,15 The comparative genomic analysis of the enterohemorrhagic E. coli strain O157:H7 and the laboratory K12 strain of E. coli has shown that nearly 30% of the genes in the pathogenic E. coli strain O157:H7 have been acquired by HGT.16 Massive archaeal-bacterial HGT has been shown between hyperthermophilic bacteria Aquifex aeolicus17 and Thermotoga maritime.18 Comparisons with mesophilic bacteria showed the presence of more ‘archaeal’ proteins in hyperthermophiles than in mesophiles.17 In another study, mesophilic archaea with relatively larger genomes, such as Methanosarcina and Halobacteria, had acquired more ‘bacterial’ genes than thermophilic archaea with smaller genomes.19–21 This implies that ~20% of the genes in an organism might have been acquired via archaeal-bacterial HGT in shared habitats.19
The presence of ten prophages and their remains in B. subtilis genome showed the evolutionary role in HGT, mainly in the propagation of bacterial pathogenesis.10 Alkaliphilic Bacillus strains cannot grow or grow poorly at neutral pH. The complete genome sequence of B. halodurans C125 has thrown light on the mechanism of adaptation of this strain to alkaline environments. The presence of a number of transposase genes (112 genes) played an evolutionary role in HGT and internal genetic rearrangement in the genome.22 The HGT in two strains of B. amyloliquefaciens (DSM7T and FZB42) has not affected the main metabolic pathways.23 The role of HGT is unclear as several of these genes are annotated as conserved hypotheticals.
Comparative Analysis of the Genomes of B. Subtilis and E. coli
The B. subtilis and E. coli genomes are similar in size. The evolutionary divergence of eubacteria into gram positive and gram negative groups has also been shown by sequencing B. subtilis and E. coli genomes. Nearly 1000 B. subtilis genes have orthologous counterparts in E. coli (one-quarter of the genome). These genes neither belong to the prophage-like regions nor to regions coding for secondary metabolism (15% of the B. subtilis genome), signifying that a huge portion of these genomes shared similar functions. One hundred putative operons or parts of the operons were conserved between E. coli and B. subtilis. Twelve of these operons showed a reshuffled gene order (typically, the arabinose operon is araABD in B. subtilis and araBAD in E. coli). Some other classes of operons, including major integrated functions like ATP synthesis (atp operon) and electron transfer (cta and qox operons), are also conserved among these microorganisms. Many operons with unknown functions are also conserved in E. coli and B. subtilis; these could be the main targets for functional analysis of these model genomes.10
Comparative Analysis of Genomes of the Genus Bacillus
The B. subtilis genome contains five signal peptidase genes along with the components of secretion apparatus which help in the secretion of macromolecular hydrolases (proteases and carbohydrases in g L–1) and antibiotics.10 Brown et al24 reported the evolution of a new strain of B. subtilis, named WN716, having different colony morphology as compared to known strains. This strain is also lacking sporulation, competence, acetoin production, motility, multiple auxotrophies, and enhanced competitive strength.25 Genome sequencing of this strain revealed no large chromosomal rearrangements, 34 single-nucleotide polymorphisms (SNPs) and +1 frameshifts. In response, many genes were affected including biosynthetic pathways, sporulation, competence and DNA repair. The whole genome sequencing within the species specified how genetic variation in the nucleotides add to the competitive robustness within species.
The genome sequence of B. subtilis natto was compared to its closely related strain B. subtilis 168. A number of insertion sequences harbored by B. subtilis natto and lacked by the latter strain, showing significant relation to natto production.24 The genome of sporogenic lactic acid bacterium B. coagulans highlights the potential of this organism to produce biocatalysts for the production of fuels and chemicals. Comparative analysis of the genome of B. coagulans with other Bacillus and Lactobacillus spp. reveals the presence of some genes common to both genera however this strain falls into the genus Bacillus. The presence of D-lactate dehydrogenase (D-LDH) encoding gene marks the potential of this organism for probiotic use in humans.26
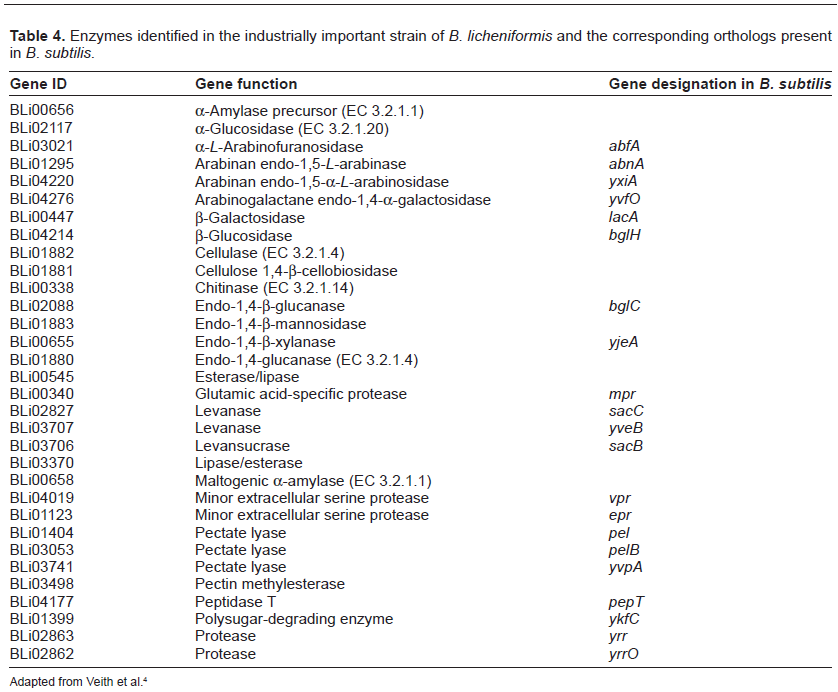
The genome of industrially important B. licheniformis DSM 13 has been studied in detail. Some industrially significant genes include those which encode carbohydrate-, lipid-, and protein-degrading enzymes. The genes having extracellular protein secretory functions are presented in Table 4. The presence of conserved regulatory DNA motives, the glyoxylate bypass, and the anaerobic ribonucleotide reductase signifies that B. licheniformis can grow on acetate and 2, 3-butanediol as well as anaerobically on glucose.11 The complete genome of B. licheniformis ATCC 14580 was sequenced and compared with closely related B. subtilis. It produces many extracellular enzymes useful in nature in nutrient cycling. The B. licheniformis and B. subtilis shared organizational similarities. However, their genomes differed in numbers and locations of prophages, transposable elements, a number of extracellular enzymes, and secondary metabolic pathway operons.27 Many genes for exoenzyme production are also encoded in the genome including protease Subtilisin Carlsberg precursor,28,29 the glutamic acid-specific protease,30,31 the maltogenic α-amylase,32 and the temperature- and pH-stable α-amylase.33–36 The analysis of the genome of industrially important alkaliphilic B. halodurans offered remarkable and significant information.37 This strain has been reported as a producer of β-galactosidase38 and xylanase.39
Enzymes identified in the industrially important strain of B. licheniformis and the corresponding orthologs present in B. subtilis.
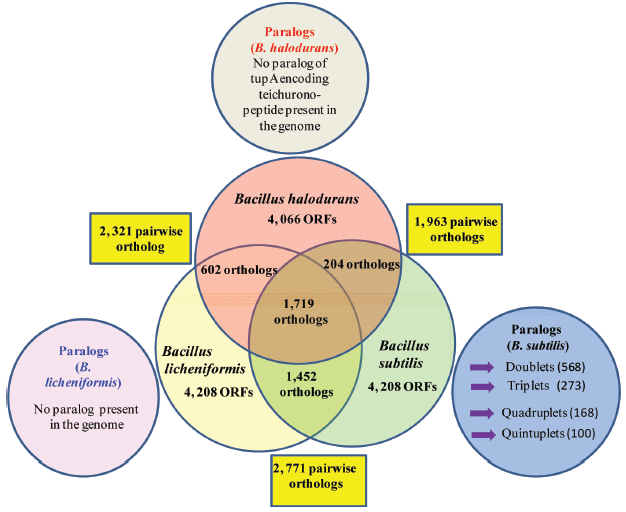
The establishment of relation between intermediary metabolism and genome structure, function, and evolution is important. Therefore, B. subtilis proteins were compared with themselves and with other complete genomes of Bacillus species. Nearly all of the essential genes in B. subtilis have orthologs in B. licheniformis, and many of them are present in a broad range of bacterial taxa. A number of paralogues are composed of big families of functionally associated proteins engaged in the transport of compounds in and out of the cell or in transcription regulation10 (Fig. 3).
Comparison of the orthologous gene complements of B. licheniformis ATCC 14580, B. subtilis 168 and B. halodurans C-125. Numbers in the rectangular boxes shows the number of pairwise orthologs between neighboring species (BLAST threshold E = 1 × 10–5). Numbers in the outer circles indicates the total number of CDSs predicted in each genome, numbers in areas of overlap represents the number of orthologs predicted by reciprocal BLASTP analysis (threshold E = 1 × 10–5), and the number in the center presents the number of orthologous sequences common to all three genomes. The outer circles show the paralogs in the genome of these Bacillus spp (Modified from Rey et al).27
B. subtilis possesses less genes involved in replication, recombination, and repair showing a relationship with the scarce repetitive elements. On the other hand, the average number of genes related to carbohydrate transport and metabolism are high, as it is a soil microbe and is in close contact with plants and their products.40 Some studies imply that the chromosome remodeling and genome reduction41–43 is not a common characteristic of B. subtilis. However, B. pumilus consists of fewer genes involved in DNA repair, oxidative stress, and acid soluble proteins that lessen the DNA damage due to desiccation and formation of UV resistant spores when compared to B. subtilis and related species.43,44
B. megaterium is a commercially available, industrially important non-pathogenic host for the production of products such as vitamin B12, penicillin acylase, and amylases. It employs Sec and Tat systems for effective secretion of proteins into the medium and accounts for a total of 30 secreted proteases genes in the genome. It is also the first biotechnological vitamin B12 producer. It is deep-rooted in the Bacillus phylogeny and is therefore significant in understanding genome evolution, dynamics, and agility in the Bacilli. The presence of an asymmetric region around the origin of replication was syntenic across the genus, a characteristic feature of the genome architecture of Bacillus spp. and sporulating lifestyle. B. megaterium is the largest of all Bacilli because of the presence of a second ftsZ gene.45 The draft genome of B. megaterium WSH-002 contains 9.26% of genes responsible for the synthesis and secretion of proteins.46
Read et al12 sequenced and analyzed the complete genome sequence of the chromosome of B. anthracis Ames for understanding anthrax pathogenesis. Many proteins that contributed to pathogenicity (haemolysins, phospholipases, and iron acquisition functions) and surface proteins, as well as the targets for vaccines and drugs have been identified. The B. anthracis genome was compared with the closely related B. cereus and B. thuringiensis which shared similarity of chromosomal genes but are not associated with anthrax. The high pathogenicity of B. anthracis H9401 is due to a tripartite exotoxin and a poly-D-glutamic acid capsule, the main virulence determinants, responsible for complete pathogenicity and are encoded by two plasmids, pXO1 and pXO2.47–49B. cereus is an opportunistic pathogen that causes food poisoning and is closely related to B. anthracis (animal and human pathogen) and B. thuringiensis (insect pathogen). Ivanova et al13 reported sequencing and analysis of B. cereus ATCC 14579 and compared it with another pathogenic strain B. anthracis A2012. The pathogenicity related genes required for invasion, establishment, and propagation of bacteria in the host are well known in B. anthracis and are also present in B. cereus, showing the similarity among the strains. Whole genome sequence of B. cereus reveals the presence of protein coding sequences (CDSs) for pathogenicity that indicates the relatedness of B. cereus to B. anthracis and B. thuringiensis. Nearly 75%–80% genes are conserved between B. cereus ATCC 14579 and B. anthracis A2012, signifying that both have a common ancestor. The analysis of the metabolic potential encoded by the sets of common genes challenges the assumption of the cereus group ancestor being a soil bacterium. The characteristic feature of soil bacteria (Streptomyces spp. or B. subtilis) is the existence of multiple carbohydrate catabolic pathways, as a variety of carbohydrates are present in the soil and plant-derived material is the major source of nutrients. A total of 41 genes for degradation of carbohydrate polymers were identified in the B. subtilis genome, in contrast to only 14 and 15 CDSs in B. cereus and B. anthracis, respectively. Furthermore, a total of 51 and 48 protease-encoding CDSs were identified in B. cereus and B. anthracis, respectively, as compared to only 30 in B. subtilis. It has been suggested on the basis of certain observations that the insect intestine may have been the natural habitat for the common ancestor of the cereus group. The major collection of antibiotic resistance genes are also the characteristic feature of B. cereus group. This underlines the probability of the cereus group ancestor as an opportunistic insect pathogen than a benign soil bacterium and the nature and origin of the B. cereus.
Shivaji et al50 have recently sequenced the complete genome of B. isronenesis isolated from an air sample collected at an altitude of 27 to 30 kilometers. As it survives in the upper atmosphere and exposed to UV radiation, it carries a uvsE gene encoding a UV DNA damage endonuclease enzyme responsible for a novel form of excision repair of DNA damaged by UV light.51 A number of antibiotic resistance proteins (penicillin acylase 2 precursor, putative niacin/nicotinamide, erythromycin esterase, putative vancomycin resistance protein, tunicamycin resistance protein, putative phosphinothricin acetyltransferase, daunorubicin/doxorubicin resistance ATP binding protein, daunorubicin, doxorubicin, and fosmidomycin resistance) are present in the genome of B. isronenesis. The genome of this novel bacterial strain can be useful in comparative genomics among phylogenetically related Bacillus isolates from different environments.
Conclusions
The genome sequences of Bacillus spp. provide a wealth of information related to gene conservation, diversity among species and systematic information, which cannot be obtained by any other approach. The understanding of the biochemistry of the organism also offers clues regarding its adaptation to the extreme environmental conditions. It can also be applied in genotype association studies, mutation screening, evolutionary studies, and environmental profiling of microorganisms. Genome sequencing has provided insights into the genetic framework of the microbial world and is a leading light of advancing technologies such as microarrays and proteomics, which have reinvigorated the field of microbiological research. There is still a need to explore new industrially important Bacillus species which possess novel genes which encode hitherto unknown products for a variety of applications.
Author Contributions
All authors reviewed and approved of the final manuscript.
Funding
Archana Sharma is grateful to the Council of Scientific and Industrial Research (CSIR), Government of India, New Delhi for awarding research fellowship while writing this review.
Competing Interests
Authors disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.
References
1.
HarwoodC.R.Bacillus subtilis and its relatives: molecular biological and industrial workhorses. Trends Biotechnol.1992; 10(7): 247–256.
2.
SatyanarayanaT., SharmaD.C., RaoJLUM, EzhilvannanM., BharadwajM., AdhikariS.Potential applications of enzymes produced by Bacillus and Geobacillus. In: SatyanarayanaT., JohriB.N., editors. Microbial Diversity: Current Perspectives and Potential Applications.IK International Pvt Ltd, New Delhi; 2005: 741–768.
3.
SatyanarayanaT., SharmaA., MehtaD.Biotechnological applications of biocatalysts from the Firmicutes Bacillus and Geobacillus species. In: SatyanarayanaT., JohriB.N., PrakashA., editors. Microorganisms in Sustainable Agriculture and Biotechnology.Springer Verlag; 2012: 343–379.
4.
NishaM., SatyanarayanaT.Thermostable archeal and bacterial pullulanases and amylopullulanses. In: SatyanarayanaT., LittlechildJ., KawarabayasiY., editors. Thermostable Microbes in Environmental and Industrial Biotechnology.Springer Verlag; 2013: 535–587.
FleischmannR.D., AdamsM.D., WhiteO.Whole genome random sequencing and assembly of Haemophilus influenzae Rd. Science.1995; 269(5223): 496–512.
7.
DanielsD.L., PlunkettG., BurlandV., BlattnerF.R.Analysis of the Escherichia coli genome: DNA sequence of the region from 84.5 to 86.5 min. Science.1992; 257(5071): 771–778.
8.
AlcarazL.D., Moreno-HagelsiebG., EguiarteL.E., SouzaV., Herrera-EstrellaL., OlmedoG.Understanding the evolutionary relationships and major traits of Bacillus through comparative genomics. BMC Genomics.2010; 11: 332.
9.
RavelJ., FraserC.M.Genomics at the genus scale. Trends Microbiol.2005; 13(3): 95–97.
10.
KunstF., OgasawaraN., MoszerI.The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature.1997; 390(6657): 249–256.
11.
VeithB., HerzbergC., SteckelS.The complete genome sequence of Bacillus licheniformis DSM13, an organism with great industrial potential. J Mol Microbiol Biotechnol.2004; 7(4): 204–211.
12.
ReadT.D., PetersonS.N., TourasseN.The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Nature.2003; 423(6935): 81–86.
13.
IvanovaN., SorokinA., AndersonI.Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature.2003; 423(6935): 87–91.
14.
OchmanH., LawrenceJ.G., GroismanE.A.Lateral gene transfer and the nature of bacterial innovation. Nature.2000; 405(6784): 299–304.
15.
OchmanH., MoranN.A.Genes lost and genes found: evolution of bacterial pathogenesis and symbiosis. Science.2001; 292(5519): 1096–1099.
AravindL., TatusovR.L., WolfY.I., WalkerD.R., KooninE.V.Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends Genet.1998; 14(11): 442–444.
18.
NelsonK.E., ClaytonR.A., GillS.R.Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature.1999; 399(6734): 323–329.
19.
KennedyS.P., NgW.V., SalzbergS.L., HoodL., DasSarmaS.Understanding the adaptation of Halobacterium species NRC-1 to its extreme environment through computational analysis of its genome sequence. Genome Res.2001; 11(10): 1641–1650.
20.
DeppenmeierU., JohannA., HartschT.The genome of Methanosarcina mazei: evidence for lateral gene transfer between bacteria and archaea. J Mol Microbiol Biotechnol.2002; 4(4): 453–461.
21.
KooninE.V., WolfY.I.Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res.2008; 36(21): 6688–6719.
22.
TakamiH., NakasoneK., TakakiY.Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucleic Acid Res.2000; 28(21): 4317–4331.
23.
RückertC., BlomJ., ChenX., RevaO., BorrissR.Genome sequence of B. amyloliquefaciens type strain DSM7T reveals differences to plantassociated B. amyloliquefaciens FZB42. J Biotechnol.2011; 155(1): 78–85.
24.
MaughanH., NicholsonW.L.Increased fitness and alteration of metabolic pathways during Bacillus subtilis evolution in the laboratory. Appl Environ Microbiol.2011; 77(12): 4105–4118.
25.
BrownC.T., FishwickL.K., ChokshiB.M.Whole-genome sequencing and phenotypic analysis of Bacillus subtilis mutants following evolution under conditions of relaxed selection for sporulation. Appl Environ Microbiol.2011; 77(19): 6867–6877.
26.
RheeM.S., MoritzB.E., XieG.Complete genome sequence of a thermotolerant sporogenic lactic acid bacterium, Bacillus coagulans strain 36D1. Stand Genomic Sci.2011; 5(3): 331–340.
27.
ReyM.W., RamaiyaP., NelsonB.A.Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillus species. Genome Biol.2004; 5(10): R77.
28.
SmithE.L., DeLangeR.J., EvansW.H., LandonM., MarklandF.S.Subtilisin Carlsberg. V. The complete sequence: Comparison with subtilisin BPN’; evolutionary relationships. J Biol Chem.1968; 243(9): 2184–2191.
29.
SyedR., WuZ.P., HogleJ.M., HilvertD.Crystal structure of selenosubtilisin at 2.0-A resolution. Biochemistry.1993; 32(24): 6157–6164.
30.
KakudoS., KikuchiN., KitadokoroK.Purification, characterization, cloning, and expression of a glutamic acid-specific protease from Bacillus licheniformis ATCC14580. J Biol Chem.1992; 267(33): 23782–23788.
31.
SvendsenI., BreddamK.Isolation and amino acid sequence of a glutamic acid specific endopeptidase from Bacillus. 1992. Eur J Biochem.1992; 204(1): 165–171.
32.
KimI.C., ChaJ.H., KimJ.R.Catalytic properties of the cloned amylase from Bacillus licheniformis. J Biol Chem.1992; 267(31): 22108–22114.
33.
GrayG.L., MainzerS.E., ReyM.W.Structural genes encoding the thermophilic α-amylases of Bacillus stearothermophilus and Bacillus licheniformis. J Bacteriol.1986; 166(2): 635–643.
34.
KandraL., GyemantG., RemenyikJ., HovanszkiG., LiptakA.Action pattern and subsite mapping of Bacillus licheniformis α-amylase (BLA) with modified maltooligosaccharide substrates. FEBS Lett.2002; 518(1-3): 79–82.
35.
StephensM.A., OrtleppS.A., OllingtonJ.F., McConnellD.J.Nucleotide sequence of the 5′ region of the Bacillus licheniformis α-amylase gene: Comparison with the B. amyloliquefaciens gene. J Bacteriol.1984; 158(1): 369–372.
36.
YuukiT., NomuraT., TezukaH.Complete nucleotide sequence of a gene coding for heat- and pH-stable α-amylase of Bacillus licheniformis: Comparison of the amino acid sequences of three bacterial liquefying α-amylases deduced from the DNA sequences. J Biochem. (Tokyo)1985; 98(5): 1147–1156.
37.
TakamiH., HorokoshiK.Analysis of the genome of an alkaliphilic Bacillus strain from an industrial point of view. Extremophiles.2000; 4: 99–108.
38.
IkuraY., HorokoshiK.Isolation and some properties of beta-galacosidaseproducing bacteria. Agric Biol Chem.1979; 43: 85–88.
39.
HondaH., KudoT., IkuraY., HorikoshiK.Two types of xylanases of alkalophilic Bacillus sp. No. C-125. Can J Microbiol.1985; 31(6): 538–542.
40.
TakamiH., TakakiY., CheeG.J.Thermoadaptation trait revealed by the genome sequence of thermophilic Geobacillus kaustophilus. Nucleic Acids Res.2004; 32(21): 6292–6303.
41.
DufresneA., GarczarekL., PartenskyF.Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol.2005; 6(2): R14.
42.
PushkerR., MiraA., Rodriguez-ValeraF.Comparative genomics of genefamily size in closely related bacteria. Genome Biol.2004; 5(4): R27.
43.
SetlowP.Spores of Bacillus subtilis: their resistance to and killing by radiation, heat and chemicals. J Appl Microbiol.2006; 101(3): 514–525.
44.
GioiaJ., YerrapragadaS., QinX.Paradoxical DNA Repair and Peroxide Resistance Gene Conservation in Bacillus pumilus SAFR-032. PLoS ONE.2007; 2(9): e928.
45.
EppingerM., BunkB., JohnsM.A.Genome sequence of the biotechnologically important Bacillus megaterium strains QM B1551 and DSM319. J Bacteriol.2011; 193(16): 4199–4213.
46.
LiuL., LiY., ZhangJ.Complete Genome Sequence of the Industrial Strain Bacillus megaterium WSH-002. J Bacteriol.2011; 193(22): 6389–6390.
ShivajiS., AraS., SinghS.K., BandiS., SinghA., PinnakaA.K.Draft genome sequence of Bacillus isronensis Strain B3W22, isolated from the upper atmosphere. J Bacteriol.2012; 194(23): 6624–6625.
51.
KannoS-I, IwaiS., TakaoM., YasuiA.Repair of apurinic/apyrimidinic sites by UV damage endonuclease; a repair protein for UV and oxidative damage. Nucleic Acids Res.1999; 27(15): 3096–3103.
52.
QuailM.A., SmithM., CouplandP.A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics.2012; 13: 341.
53.
GengW., CaoM., SongC.Complete genome sequence of Bacillus amyloliquefaciens LL3, which exhibits glutamic acid-independent production of poly-γ-glutamic acid. J Bacteriol.2011; 193(13): 3393–3394.
54.
ChunJ.H., HongK.J., ChaS.H.Complete genome sequence of Bacillus anthracis H9401, an isolate from a Korean patient with Anthrax. J Bacteriol.2012; 194(15): 4116–4117.
55.
DengY., ZhuY., WangP.Complete genome sequence of Bacillus subtilis BSn5, an endophytic bacterium of Amorphophallus konjac with antimicrobial activity for the plant pathogen Erwinia carotovora subsp. carotovora. J Bacteriol.2011; 193(8): 2070–2071.
56.
ZeiglerD.R.The genome sequence of Bacillus subtilis subsp. spizizenii W23: insights into speciation within the B. subtilis complex and into the history of B. subtilis genetics. Microbiology.2011; 157(Pt 7): 2033–2041.