
Abstract
Aspergillus species are industrially and agriculturally important as fermentors and as producers of various secondary metabolites. Among them, fungal polyketides such as lovastatin and melanin are considered a gold mine for bioactive compounds. We used a phylogenomic approach to investigate the distribution of iterative polyketide synthases (PKS) in eight sequenced Aspergilli and classified over 250 fungal genes. Their genealogy by the conserved ketosynthase (KS) domain revealed three large groups of nonreducing PKS, one group inside bacterial PKS, and more than 9 small groups of reducing PKS. Polyphyly of nonribosomal peptide synthase (NRPS)-PKS genes raised questions regarding the recruitment of the elegant conjugation machinery. High rates of gene duplication and divergence were frequent. All data are accessible through our web database at http://metabolomics.jp/wiki/Category:PK.
Background
Polyketide synthase (PKS) genes generate a class of structurally diverse products; they are found in all kingdoms except archaea, which do not synthesize fatty acids. 1 While it is difficult to elucidate the biosynthetic mechanisms of each polyketide from only genomic sequences, the recognition of PKS genes is rather straightforward. They exhibit a pronounced domain architecture in common with fatty acyl synthase (FAS) genes, ie, three essential domains (ketosynthase (KS), acyltransferase (AT), and acyl carrier protein (ACP)), and optional tailoring domains such as ketoreductase (KR)-, dehydratase (DH)-, enoylreductase (ER)-, methyltransferase (ME)-, and thioesterase (TE) domains. Based on the domain architecture, research on PKS genes developed the type I, II and III paradigm.2,3 Type I, resembling yeast and animal FAS, is a large multi-domain enzyme that produces a variety of structures (details follow). Type II, resembling plant and bacterial FAS, is a single-module enzyme lacking the AT domain. In vivo, type II enzymes are thought to form multi-enzyme complexes to synthesize aromatic compounds. Type III, known as ‘chalcone synthase-like’, is mainly associated with plants and structurally and mechanistically distinct from type I and II; it does not contain the otherwise essential ACP domain. In this work we focus on type I iterative genes, mostly in fungi but also in other kingdoms.
Type I genes are at least 5 kbp in length and they harbor multiple catalytic domains. Their two classes,
In fungi, on the other hand, the same domains are iteratively used in the course of chemical condensation. Such genes are designated type I iterative PKS (iPKS) genes. The control mechanisms that determine the number of iterations are not fully understood. 4 Depending on the reduction degree of their final product, type I iterative genes are further classified into three categories, ie, non-reducing-, partially reducing-, and reducing PKS (NR-, PR-, and R-PKS, respectively). Fully reduced polyketides correspond to fatty acid derivatives; this also indicates the relationship between PKS and FAS.
The availability of fully-sequenced genomes provides a new avenue for the study of the evolutionary relationship and diversity of PKS genes in different organisms. Some fungi have type I modular genes that produce diketides and some bacteria possess type I iterative genes.5,6 The existence of nonribosomal peptide synthase (NRPS) genes further complicates the species-metabolite distribution. NRPS genes are megasynthases similar to PKS genes. When a PKS is adjacent to a NRPS, the final product may be a hybrid of polyketide and small peptides. A good example is the mycotoxin alpha-cyclopiazonic acid in
The full genome sequences of eight
We created a web database to explore and understand the diversity of iPKS genes in
Methods
PKS genes and genome databases
We collected the amino acid sequences in the GenBank Database for eight
Throughout our study we referenced several PKS-specific databases for domain information. For functionally identified PKS genes we used PKSDB and ITERDB (National Institute of Immunology, India), which contain information and references on 20 modularly- and 13 iteratively synthesized polyketides, respectively.22,23 The other information source was MapsiDB developed in Korea by SmallSoft Co., Ltd. 24 This database provides genomic information on 45 modularly and 21 iteratively synthesized polyketides and is part of the MAPSI (Management and Analysis for Polyketide Synthase Type I) prediction system that is based on the hidden Markov model. We used this tool extensively in our domain predictions.
Assignment of catalytic domains and PKS types
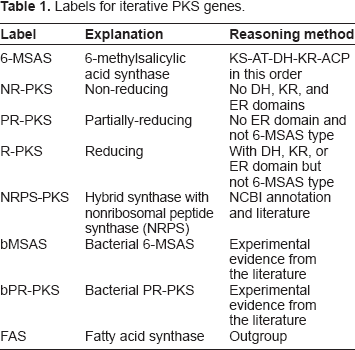
Amino acid sequences were analyzed with MAPSI and the Conserved Domain Database (CDD) from the National Center for Biotechnology Information (NCBI). 25 The use of multiple prediction tools is important because MAPSI recognizes neither the ME domain nor domains in NRPS genes. CDD, a general-purpose system not optimized for PKS, is based on a position-specific scoring matrix and yields many false positives. We manually removed unreasonable predictions such as domains unrelated to PKS. After determining the domain composition we assigned each gene to one of 7 PKS types (Table 1). Labels for NRPS-PKS and bacterial iPKS (bMSAS or bPR-PKS) were assigned only when such assignment was supported by the NCBI web resource (especially GenBank) or other literature sources. The remaining 4 types (6-MSAS, NR-PKS, PR-PKS, or R-PKS) were assigned based on literature information or the domain composition predicted by computational tools. Thus, we first assigned the 6-MSAS label to small genes with KS-AT-DH-KR-ACP domains in this order.15–16 Next, the NR-PKS label was assigned to genes without any DH, KR, and ER domains. We assigned the PR-PKS label to genes without the ER domain and the R-PKS label to the remaining genes. Whenever gene functions were experimentally identified we assigned labels according to the biosynthetic role of the genes. Fewer than 20% of all PKS genes, however, have been functionally identified by detailed experiments.
Labels for iterative PKS genes.
Domain genealogy construction
According to our data curation policy, all genes must contain both KS and AT domains. Multiple alignments of amino acid sequences were performed by CLUSTALW and MUSCLE software embedded in the MEGA program package (Version 5.0) with manual corrections. 26 We did not use sequences with incomplete domains. Phylogenetic inference was obtained with neighbor-joining (NJ)- and maximum-parsimony (MP) algorithms with a bootstrap test (1000 pseudo-replicates) in the MEGA package. Visualization was with the TreeDyn software package. 27
The concise KS tree was created for a reduced set of domain sequences. After generating the complete tree with all KS domains we one-by-one chose reliable clades with a bootstrap value greater than 75 and fed them into the CD-HIT program to obtain representative sequences for each clade. 28 Lastly, the chosen sequences were subjected to the phylogenetic algorithms to form a reduced tree.
Database construction
PKS genes from
Results and Discussion
Distribution of PKS-related genes
In total, we collected 400 type I iPKS-related genes from fungi and 71 from bacteria. To our knowledge, our collection is the largest publicly accessible, manually curated data resource focusing on iPKS in
The statistics of the iPKS genes for the eight
The number of PKS genes in sequenced

Obligate plant pathogens such as
The numbers in Table 2 are not definitive. The statistics are intended as upper bounds and may change with the counting method used. For example, for
Domain compositions
Domains were manually assigned by referencing computational predictions such as MAPSI, ITERDB, and CDD as described in the Methods section. The MAPSI tool can assign 7 domains (KS, AT, ACP, KR, DH, ER, and CYC) and CDD can assign more than 20 different domains. Information on predicted domains for all sequences is freely accessible through our wiki-based database at http://metabolomics.jp/wiki/Category:PK by following links at the top of the page. The classification of PKS types was based on the literature and on the database annotation as described (Table 1). The 6-MSAS type was assigned for small genes with the domain structure KS-AT-DH-KR-ACP.15,16 Although the assignment may look simplistic, our phylogenetic analysis supports this definition (see the concise phylogenetic trees shown in Figs. 1 and 2). The remaining iPKS genes were assigned to the nonreducing (NR)-, partially reducing (PR)-, or reducing (R) type depending on the existence of reducing domains and literature information, if any (see Methods). Since the number of functionally identified genes is small, their categorization largely depends on the predictions by the MAPSI tool which is specialized for type I PKS. We listed results of both MAPSI and CDD in our website entries and added more entries if literature information was available. The statistics of the MAPSI output are shown in Fig. 3. Among the 7 assigned domains, KS was the longest (422 aa ± 21 SD), followed by ER (320 aa ± 9 SD), and AT (300 aa ± 13 SD).Thenumber of the inessential ER domain is only one quarter of KS. Some reducing PKS genes in bacteria did not have an ER domain; such exceptions were rare in fungi.

The phylogenetic tree of the KS domain for representative sequences (ie, the concise tree) computed with the maximum parsimony method.

Overview of the KS domain phylogeny.

Statistics of the number of domains and their length predicted by the MAPSI tool.
The statistics of the domain length justify our decision to consider only sequences of more than 800 aa as residues for PKS. In fact, the CADRE database contains shorter PKS genes, but their domains are partial and probably not functional. Such genes were excluded from our analysis and from Table 2.
Since some PKS genes exploit external genes, the assigned types may not always correspond to the chemical type of their products. For example, the lovastatin synthase in
Phylogenomic analysis of KS domains
The KS region is the best conserved domain. Phylogenetic estimates on KS only coincide with results that are based on combined KS and AT domains. The only exceptions are PKS genes from the protozoa
Phylogenetically, NR-PKS, (P)R-PKS, and 6-MSAS types clustered well. 38 The NR-PKS type is adjacent to bacterial PKS genes, many of which are involved in phenolpthiocerol synthesis. There are two fungal clades deep inside bacterial PKS (called the nested fungal clade or NFC); one is for NRPS-PKS and the other, larger one, for 6-MSAS. The two NFCs were statistically supported to have originated in acti-nobacteria and to have been horizontally transferred later into ascomycete fungi.16,38,41
The NR-PKS type is separated into three classes (I–III in Fig. 1). The NR-PKS I type contains genes for aflatoxins, melanins, pigments, and naphthopyrones. NR-PKS III type contains one gene for citrinin (pksCT in
In contrast to the well-clustered NR-PKS genes, R-PKS and 6-MSAS types were separated into many small groups. The R-PKS type contains the genes for fumonisin, lovastatin, and t-toxin but few genes have been experimentally verified. A recent excellent work identified a number of new products for NR-PKS genes in
Orthologous genes
Phylogenetic analysis yields information on orthologous genes. We used ‘orthology’ loosely to describe genes with identical domain structures with highly similar KS sequences. Such orthologs are expected to synthesize polyketides with identical backbones, although their structural modifications may vary depending on the external modifier genes or ecological usage. Surprisingly, only two orthologous genes were conserved by all sequenced
Well conserved orthologous genes in

Flexible rearrangements are also evidenced upon comparison of close pairs, ie,
Web-based database system
When sequences of more than 400 genes are involved, it is difficult to obtain detailed information for each sequence from published research only. Most genomic information is available on the Internet, eg, from NCBI and EBI repositories. Thus, a web-based database is the best interface to fully utilize the results of phylogenomic analyses. We created a website for PKS information in
Funding Sources
This research was supported by a MEXT Grant-in-Aid for Scientific Research on Innovative Areas “Biosynthetic Machinery” 11001359. The early part of the research was carried out at The University of Tokyo when SHL was a visiting student in Japan. His sojourn was financially supported by the National Security Council of Taiwan under Grant No. NSC096-2917-I-007-005 and by the Elite Scholarship Program, Ministry of Education, Taiwan.
Competing Interests
MA's institution received a grant for JST-NSF joint research on metabolomics. Other authors disclose no potential conflicts of interest.
Author Contributions
Conceived and designed the experiments: SHL, MA. Analyzed the data: SHL, MY, MA. Supervised the data analysis of SHL: PCL, CYT, MA. Wrote the first draft of the manuscript: SHL, MA. Agreed with manuscript results and conclusions: SHL, PCL, CYT, MA. Made critical revisions and approved final version: SHL, MA. All authors reviewed and approved the final manuscript.
Supplementary Data
Supplementary Figure SI: Attached as a separate file. Supplementary Sequence Data for 471 genes: Information on predicted domains and species.
Supplementary FASTA format of the 43 KS clusters: Information on aligned groups generated by the Geneious software program (basic version v5.63).
Supplementary Figure

Footnotes
Acknowledgements
We thank Prof. Isao Fujii (Iwate Medical University, Japan) for comments and kind suggestions on our manuscript. We also thank Ms. Ursula Petralia for editing our manuscript.
As a requirement of publication, the author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.