
Abstract
Advances in bioinformatics and sequencing technologies have allowed for the analysis of complex microbial communities at an unprecedented rate. While much focus is often placed on the cellular members of these communities, viruses play a pivotal role, particularly bacteria-infecting viruses (bacteriophages); phages mediate global biogeochemical processes and drive microbial evolution through bacterial grazing and horizontal gene transfer. Despite their importance and ubiquity in nature, very little is known about the diversity and structure of viral communities. Though the need for culture-based methods for viral identification has been somewhat circumvented through metagenomic techniques, the analysis of metaviromic data is marred with many unique issues. In this review, we examine the current bioinformatic approaches for metavirome analyses and the inherent challenges facing the field as illustrated by the ongoing efforts in the exploration of freshwater phage populations.
Introduction
Advances in sequencing technologies over the past two decades have led to many studies examining the complex microbial communities that support life on Earth. From extreme environments 1 to buildings, 2 surveys of microbial communities have identified many novel taxa of bacteria and archaea.3,4 In parallel, exploration of the viral fraction of microbial communities has also consistently uncovered novel genetic content.5,6 With an estimated 10 30 viruses present in the ocean alone, 7 and considering our current surfeit of knowledge, 8 viruses represent a vast untapped reservoir of genetic diversity.9,10 Viruses that infect bacteria (bacteriophages) are some of the most abundant biological entities on the planet: they play a critical role in shaping microbial community structure and metabolism through mediation of mortality and genetic mobility. 11 Therefore, phages have a global impact on biogeochemical nutrient cycling. 12
Assessing the diversity of viral species is not as straightforward as it is for prokaryotes. While bacteria and archaea can be classified via the 16S rRNA gene, there is no single conserved gene among all viral species. Thus, whole-genome sequencing (WGS), rather than targeted gene sequencing, must be employed to begin to assess the heterogeneity of viral taxa present within a sample (the metavirome). The WGS approach has also been employed in numerous studies of bacterial and archaeal communities13–15; these studies have a crucial advantage – a well-populated repository of characterized gene sequences. Despite the critical role phages play in the natural environment as well as within the human microbiota,7,16–21 data repositories lack sufficient quantity and diversity of phage gene and genome sequences. This is due to a general dearth of phage research in comparison to that of all other organisms, as well as the challenges associated with working with phages in the laboratory. Predominantly, the isolation of phages is limited in the same way as that of bacteria – only a fraction can be successfully isolated from the environment and maintained in laboratory settings. In addition, direct isolation and the use of metagenomics-based analyses are heavily biased in favor of examining the communities of phages that are actively destroying bacterial cells. These lytic phages are present as unattached viral particles in the environment. However, a large cohort of phages integrate with host genomes in a state known as lysogeny, forming prophages. Prophages are often overlooked during metaviromic examinations, along with all other bacterial data. Furthermore, many prophages are mislabeled in data repositories as being innately bacterial.22,23
Interest in phage ecology and community dynamics within natural and man-made environments continues to accelerate. 24 Herein, we review the current bioinformatic approach for metavirome analyses with specific focus on the study of complex phage communities. While phage communities have been investigated in a variety of ecosystems, we focus on phage communities in freshwaters. Freshwater represents a potentially highly dynamic and variable community of phages and is likely to support a novel cohort of genetic and phenotypic diversity. In comparison to the marine environment, freshwater phages have been understudied: in light of the limitations already discussed, this paucity of data contributes to our currently restricted insight into these important communities. Such hindrances reflect the general technicalities that are associated with the analysis of metagenomic data, which are, inarguably, very valuable to our broader knowledge of viral and microbial communities in the environment. Herein, we begin to examine the practical aspects of bioinformatics-based analyses of metaviromic data, particularly those from freshwater, and how solutions to associated problems may inform the broader field.
Bioinformatic Analysis of Environmental Metaviromic Datasets
Current methodology
It is common practice to follow the well-established protocols developed for the investigation of microbial communities. 25 Prior to beginning analyses, raw viral sequencing reads should be inspected to remove sequencing artifacts (eg, primers and/or adaptors). While contemporary sequencing platforms typically have very low overall error rates, errors and biases do occur 26 and can be identified and removed alongside low-quality bases via a number of tools [eg, FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and the FASTX-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/)]. Given the importance of high-quality data, several open-source software tools have been developed to facilitate quality control (eg, HTQC 27 ; SUGAR 28 ; khmer 29 ).
While there are many methods for processing WGS datasets, extant metaviromic studies – particularly those focused on complex phage communities – typically follow a protocol similar to that shown in Figure 1. The reads produced, irrespective of the sequencing technology used, are first assembled into contigs using tools such as Velvet 30 and SPAdes. 31 While Figure 1 lists some of the assemblers frequently used within freshwater metavirome studies, this is in no way an exhaustive representation of the tools available.25,32,33 Postassembly, the contigs can either be directly compared with viral data collections or open reading frame (ORF) prediction can be performed. In the latter case, several different software tools are publicly available, including those listed in Figure 1. Contigs are classified through heuristic homology comparisons (typically BLAST 34 or in some cases BLAT 35 ) to available viral sequence collections, such as GenBank, 36 RefSeq, 37 and SEED. 38 Downstream analyses and comparisons between viromes often rely on these homology-based results.
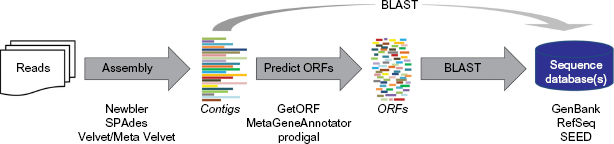
General protocol for metaviromic data analyses. Commonly used resources are listed for individual steps.
There are several cloud-based or remote services, which encapsulate the process outlined in Figure 1. While both MG-RAST 39 and MEGAN 40 were developed for analyses of bacterial community sequencing efforts, they can be applied to metavirome data analyses. Two tools, MetaVir 41 and VIROME, 42 have been designed specifically for the analysis of metaviromic data sets. Sequences are classified by MetaVir through the top blastx comparisons to the RefSeq Virus database. 41 VIROME classifies viral sequences via the top blastp comparisons to the UniRef100 peptide database. 42
Limitations of mainstream methodologies
The ability to classify a sequence originating from a phage, whether attempting to identify the taxa present or the putative functionality of a coding region, is dependent upon the availability of representative sequences within the data repository used. As of January 2016, EBI's Phage Genome collection (www.ebi.ac.uk/genomes/phage.html) includes only 2,010 organisms, similar to that reported in the RefSeq collection through NCBI (2,018 total). NCBI's Nucleotide database (www.ncbi.nlm.nih.gov/nuccore) has just over 12,000 phage sequences (complete and partial), nearly three orders of magnitude less than the size of bacterial sequence collections. Thus, analyses of metaviromic datasets must acknowledge the dependency upon available sequence databases and the limited number of characterized species. The small fraction of phage diversity that has been sequenced also has inherent bias. dsDNA phage genomes outnumber those of RNA genomes, with Mycobacteriophages being the most expansively investigated, largely due to the work of PhageHunters (Fig. 2). 43 Likewise, phages that infect the commonly encountered type strain hosts (ie, Pseudomonas and members of Enterobacteri-aceae) are also comparatively overrepresented (Fig. 2). This imbalance within the databases presents a challenge when assigning importance to previously unclassified viral sequences from uncultured viral samples.44,45

Composition of the annotated host for the current collection of complete phage genomes available. Other taxa include all genera/families not listed here.
Furthermore, many sequence annotations are incorrect. Incomplete or mislabeled viral sequences in public databases are common. For instance, a blastx search of nucleic acid from purified virus-like particles against GenBank was shown to produce matches to nonviral sources. 46 However, when these same sequences were compared with the ACLAME database (a collection of classified mobile genetic elements including phages, plasmids, and transposons 47 ), the majority of reads could be reclassified as plasmid or phage. 46 Two tools have been developed to assist in identifying sequences of viral origin within microbial genomes: VirSorter 48 and PhagePhisher. 49 Both can identify lysogenic viral sequences as well as prophage sequences. Prophages within bacterial genomic sequences can lead to erroneous classification of metaviromic sequences as being bacterial in origin, and thus necessitates further, often manual, investigation of such hits. Limiting comparisons to only annotated viral sequences circumvents this challenge; however, this comes at a cost of ignoring phage sequences that have only been identified within bacterial genomes (ie, their species of origin has yet to be discovered and characterized).
In addition to practical aspects of computational analyses, bias is also introduced during isolation and sample preparation. Filtering, a standard aspect of water sample preparation, 50 potentially excludes large dsDNA viruses.44,51,52 Additionally, chloroform treatment, CsCl gradients, and random PCR amplification favor chloroform-tolerant viruses, tailed phages, and abundant taxa, respectively. 53 Amplification of whole DNA via techniques such as multiple displacement amplification is known to favor single-stranded circular DNA in the amplification process, 54 which may result in the overrepresentation of these viruses in some metaviromic studies. Sample storage times and temperatures may also exclude some environmental viruses as their decay rates vary. 44
Exploring metaviromes – case study: freshwater samples
The initial discovery that phages were highly abundant in aquatic samples 55 paved the way for the eventual determination of the pivotal impact that phages have on global bacterial-mediated processes. Phage metagenomics essentially began in the marine environment, 56 and marine datasets continue to be one of the most comprehensively examined from a biological and bioinformatic perspective (eg, The Pacific Ocean Virome 10 ). There are comparatively few datasets collected from freshwaters, despite their importance as sources of drinking water, recreation, and commerce. At the microbial scale, water chemistry and hydrological factors can contribute to a dynamic environment, which is likely to be reflected in the indigenous phage populations.
To date, there have been 12 freshwater metavirome datasets, which focus on phage populations within their samples and for which details regarding their methods and results of analyses are published (Table 1 and Fig. 3). Many of these studies have been performed using 454 sequencing, as presented in Table 1. In addition to the studies listed in Table 1, there have been several other metavirome datasets generated. A search through, eg, NCBI's Sequence Read Archive (SRA: http://www.ncbi.nlm.nih.gov/sra/) or the iMicrobe data commons (http://data.imicrobe.us/), will reveal additional sequencing datasets from various other freshwater environments. Given our motivation here is on the analyses of metaviromic datasets, we will thus focus on those studies accompanied by publications.
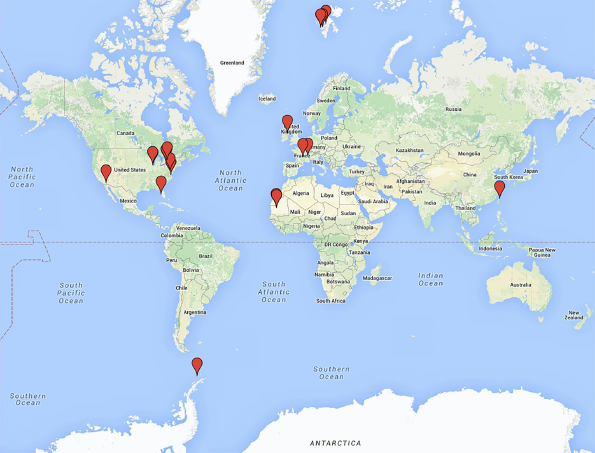
Locations of the freshwater metaviromes in Table 1.
Published freshwater metavirome studies of phages.

As presented in Table 1, the majority of the sequences generated by metaviromic studies exhibit no discernible similarity to the sequences in current data repositories; in fact, at most 29.5% of the sequences generated exhibit homology to a known viral sequence. This is universal, regardless of the freshwater ecosystem under investigation. Freshwater metavirome surveys have largely been focused on DNA viruses, which include nucleic acids from both phages and eukaryotic viruses. However, phages constitute the majority of the identifiable sequences within these datasets.51,57–59 Nevertheless, from the small fraction which is identifiable, varying conclusions have been drawn.
Nutrient levels affect phage community structure
Previous work suggests that the diversity of DNA phages in freshwater environments is subject to change based on various environmental factors including temperature and available nutrient levels.45,60 Freshwaters of lower nutrient availability tend to have less viral species richness in comparison to their higher trophic counterparts, eg, the mesotrophic Lake Bourget and oligotrophic Lake Pavin. 60 This corresponds to greater taxonomic diversity of bacterial hosts within environments with high nutrient availability. However, this contradicts a study performed on Antarctic samples from the oligotrophic Lake Limnopolar, which demonstrates high species richness. 45 Given the fact that the databases, from which species identification is made, are themselves not representative of phage diversity, claims of changes in diversity are inherently biased. Furthermore, as the vast majority of the virome is unidentifiable, quantifying the true diversity present is not possible.
Variation due to seasonality and weather
Correlations between season and species diversity have also been observed.45,58 Furthermore, shifts in the taxa have been associated with environmental stressors, including decreased rainfall. 57 In a study examining Saharan gueltas (ponds), the authors postulated that the extreme conditions of this site favor lysogenic phages, supported by their observation of Microbacterium phage Min1. 57 Given phage dependence upon the presence and susceptibility of their bacterial host(s) and the seasonality previously observed within bacterial species in freshwaters,61,62 fluctuations in viral population structure are expected. These results rely on the small fraction of identifiable sequences. Exploring the unknown constituent is far more difficult. Cross-sample assemblies – assembling contigs from one sample with another sample – can provide some insight into the similarity/dissimilarity between sequences, regardless of their homology to databases.44,63
Spatial and temporal variation
Examination of freshwater bodies from the Arctic and Antarctic revealed similar taxa within their waters, despite their geographic distance.45,64 Similarly, when the metaviromes of Lake Ontario and Lake Erie were examined concurrently, little to no significant difference in species diversity was observed. 51 However, temporal variation was observed in both lakes when taxonomic composition was examined between samples taken a year apart. For example, although dominant in the collections of 2012, in 2013 Myoviridae populations decreased in both Lake Ontario and Lake Erie samples. 51 While these two studies compare somewhat similar habitats, comparison between diverse environments (such as those presented in Table 1) highlights the variability observed. Nevertheless, the same caveats discussed earlier are just as true here.
Anthropogenic effects
The effects of human-related impact on the microbial environment have recently been attracting more attention. In a study of the Virginian Lake Matoaka, viral species richness and diversity was found to be negatively correlated with the level of human activity at the sample site, with the highest levels of diversity and species richness found at the main body of the lake, 54 the area least affected by human activity. Similar results were found in Saharan gueltas 57 ; the guelta most influenced by human beings exhibited the lowest amount of viral diversity and more heterotrophic microorganisms and human pathogens. When potable and reclaimed water were compared in terms of viral abundance, it was found that the latter contained 1,000-fold more viruses than the former 46 ; reintroducing reclaimed water within the environment can thus potentially affect native microbial species.
Caveats of Applying Existing Methods of Analysis to Environmental Metaviromic Datasets
As the discussed studies of freshwater phage populations exemplify, the dearth of characterized phage genome sequences limits the ability to classify the majority of metaviromic sequences. The genomes of phages are extremely plastic and are able to shuttle genes between organisms 65 ; looking at the number of BLAST hits to a single genome may erroneously indicate the species' presence. For instance, several thousand hits to the Planktothrix phage PaV-LD genome suggested the presence of this phage within the Lake Michigan nearshore waters. 66 However, further investigation of these BLAST results revealed that these hits were to a single gene, thus suggesting that the gene rather than the species was present within the sample. 66 Thus, BLAST is an effective means of assessing the presence and absence of genes of interest; similarly, metaviromic sequences could be mapped to user-selected gene sequences to detect and qualify the prevalence of genes of interest. Nevertheless, in the absence of a conserved genetic marker, ascertaining the presence of particular species within metaviromic sequences is fraught with challenges that have yet to be addressed.
As metaviromic surveys of environments continue, it is imperative – regardless of the environment sampled – that any conclusions drawn with regard to incidence of particular viral species be informed, considering not only the number of hits to a particular reference genome but also its coverage (the percentage of a query sequence that is aligned with a database sequence). Samples that produce hits to the majority of a genome's coding regions may signify the abundance of a particular taxon. The complete genomes of highly prevalent species have been successfully constructed from complex metagenomic datasets, eg, the highly abundant crAssphage within fecal samples. 67 Mapping reads to a reference genome of interest can also be used to classify and extract complete genome sequences. While neither approach has to date been applied to phage genome reconstruction within freshwater samples, some success has been possible with samples from other environments.68,69 The genomic plasticity of phages again presents a challenge as gene order and content between a reference genome and an isolate from nature may vary significantly, thus limiting the effectiveness of mapping strategies.
Furthermore, hypothesis-based inquiry of complex viral communities in nature is limited by the quality and quantity of data available. In an effort to gain insight into the putative predator-prey dynamics of phage and host, a metaviromic survey 66 in parallel with a bacterial 16S rRNA gene survey 70 of Lake Michigan nearshore waters was conducted. Metaviromic sequences were examined following the procedure outlined in Figure 1 (Velvet → getOrf → BLAST against NCBI's viral RefSeq database). Metavirome contigs were classified via MetaVir 41 ; for each phage species detected, its annotated bacterial host was determined referencing the NCBI genome record and the associated literature. Figure 4 illustrates the observed (via 16S rRNA gene sequencing) relative abundances of bacterial phyla vs. the expected (via annotated host species for phage identified) for eight samples collected from one of the two sites along the Chicago shore of Lake Michigan. As Figure 4 shows, the majority of the phage species identified within the metaviromic analyses are documented for a host species that were not detected by the bacterial survey. Highly prevalent phage species included Pseudomonas-infecting species; however, pseudomonads were found in very low abundance based on the 16S rRNA gene survey. Most notably, Figure 4 suggests a large contingent of phages, which infect other bacterial taxa, predominately cyanobacteria; however, cyanobacteria were not detected via the complementary 16S rRNA gene survey.

Phage-host populations within Lake Michigan nearshore waters.
The observed phage diversity reflects the underlying database (correlation of a number of predicted hosts to that of the annotated hosts for the available RefSeq phage sequences is r 2 > 0.99). While many factors (limited available data, misannotated and/or incomplete annotation of phage host species, etc.) may contribute to this discord, Figure 4 highlights the fact that without improved phage data resources, high-throughput sequencing projects are likely to uncover artifacts of the data repositories themselves. In many cases, the annotated host species for a phage genome is limited to a single, often laboratory, bacteria strain. Broad host range phages – phages capable of infecting bacteria belonging to the phyla Proteobacteria, Actinobacteria, and Bacteroidetes – have been isolated from Lake Michigan. 71 This generalist lifestyle has benefits within environments for which bacterial communities, ie, susceptible host populations, are often ephemeral. It is important to note that the Lake Michigan broad host range viruses are relatives of the Pseudomonas PB1 phage, which to date has only been documented as infecting Pseudomonas spp. 72 This suggests that phage species within the annotated collection of sequences may in fact have a broader host range than currently documented.
Moving Forward
In this review, we have illustrated the bioinformatic challenges faced by all metaviromic studies. These include poor representation of most virus groups in sequence repositories relative to their expected diversity in nature; overrepresentation of viruses infecting a handful of model laboratory bacterial strains; low rates of isolation and characterization of new phages in the laboratory; and generally, poor annotation of the genomes for phage that have been sequenced. Furthermore, while we have focused largely on dsDNA phages, RNA and ssDNA phages are important members of viral communities. 73 While a few studies have been conducted,45,74,75 this fraction of the virome often goes unsequenced and unsampled due to additional difficulties with their genomic extraction and amplification. If the goal is to obtain a fair representation of all viruses in data repositories, then more work is also needed to identify the non-dsDNA phages. Even when sequenced, RNA phages can be especially difficult to identify, either due to their relative scarcity in a particular sample 76 or due to biases in the analysis itself. 77 Nevertheless, the bioinformatic obstacles highlighted here also extend to non-dsDNA phages. We have learned a great deal from metaviromic studies across biomes, and there are several steps available to improve the state of metaviromic bioinformatics going forward. We emphasize the role that freshwater studies can play in leading the way.
First, more studies of freshwater phages will help to improve the relative sampling of virus groups in sequence databases. To date, most studies have focused on phages from soil,78–80 sewage, 81 and marine environments,10,56,82–84 with only a handful of studies reporting on the diversity of phages in freshwater (including those listed in Table 1). This is particularly surprising, as freshwater rivers and lakes have a direct impact on society and human health. Furthermore, freshwater ecosystems are available throughout the world at different latitudes and altitudes and include bodies of water of varying size, productivity, and geological history. Taken together with the seasonal dynamics of many freshwater systems, we expect these environments to harbor a great deal of undiscovered viral diversity.
Second, it is necessary to isolate and characterize phages uncovered from new metaviromic studies. Thus, virus groups in sequence repositories will be better represented aiding in the analyses of environmental viromes. Of central importance is the comprehensive characterization of new isolates. This goes beyond sequencing genomes; assaying growth characteristics (eg, latent period, generation time, and burst size) and the phage's host range (following protocols such as Ref. 85) are imperative to furthering our understanding of environmental phages. In doing so, it will also be important to move beyond isolation on typical laboratory hosts and to include coisolated bacterial species. More work is also needed to characterize viral gene functions. Even for one of the best-studied laboratory strains, Enterobacteriophage T4, 114 of its 278 genes are currently annotated as hypothetical proteins in GenBank. Sequencing more genomes alone cannot fill in these gaps in our knowledge. RNA-seq has been used to track bacteriophage gene expression in vivo (eg, in the oral microbiome 86 ), and similar approaches may also help to identify important, unannotated genes, in viral communities.
Third, new analytical tools are necessary to make sense of the data that we do have. While the studies reviewed here often follow a bioinformatic analysis strategy akin to that presented in Figure 1, alternative approaches developed for prokaryotic and eukaryotic metagenomic studies (eg, those reviewed in the studies by Li and Homer 32 and Nagarajan and Pop 33 ) may be applicable as well. Such methods require thorough vetting using both synthetic and real metavirome datasets. Furthermore, existing computational tools for protein structural and functional prediction may be adapted to the task of high-throughput prediction of novel functions in viral communities. New tools may also predict virus host range from genome sequences alone. Existing methods have recently been reviewed 87 and it is found that simple methods, such as finding homologous genes in phage and bacteria using BLAST, can accurately predict many phage hosts. Though the best success rate of these current approaches is only near 40%, these tools provide an optimistic starting point for developing new methods. Improving host prediction from metagenomes would provide database-free analysis of local virus and bacterial interactions.
Freshwater systems, particularly freshwater lakes, offer unique opportunities for hypothesis-driven metaviromics that is to varying degrees independent of these database limitations. As discussed above, previous studies have explored how nutrient availability, seasonality, temperature, and human activity influence freshwater viral communities. Experimental metaviromics controlling for these variables should play an exciting role in both developing new computational tools and exploring viral ecology and evolution.
Footnotes
Acknowledgments
The authors thank the past and current members of the Putonti Lab who have assisted in collecting samples for the metaviromes generated by our group.
Author Contributions
Conceived and designed the study: JWS, SCW, CP. Collated and conducted the analysis of publicly available freshwater metavirome data sets: KB, KM, AC, ES. Wrote the first draft of the article: KB, KM, AC, ES. All the authors reviewed and approved the final article.