
Abstract
Land plants are a natural group, and Charophyte algae are the closest lineages of land plants and have six morphologically diverged groups. The conjugating green algae (Zygnematales) are now suggested to be the extant sister group to land plants, providing the novel understanding for character evolution and early multicellular innovations in land plants. We review recent molecular phylogenetic work on the origin of land plants and discuss some future directions in phylogenomic analyses.
Introduction
The colonization of land by plants was a major event in plant evolution, transforming the environment on land.1,2 Knowledge of the origin of land plants is a prerequisite for understanding the transition from the aquatic to the terrestrial habitat of plants. The green algae are basically divided into Charophyte and Chlorophyte algae, and it is agreed that the Charophyte algae are the closest algal relatives of land plants. 3 Analyses of both morphological and molecular data have established that land plants evolved within Charophyte algae more than 450 million years ago.4,5 The Charophyte algae are mostly freshwater green algae with diverse morphologies, comprising six distinct groups: Mesostigmatales, Chloroky-bales, Klebsormidiales, Charales, Coleochaetales, and Zygne-matales. Of these, the latter three (Charales, Coleochaetales, and Zygnematales) have been considered the ancestors of land plants (Fig. 1). However, which group of Charophyte algae is most closely related to land plants has remained controversial over the past decade. In recent years, large amounts of molecular data are available and methodological developments are increasing at a fast pace, thus investigating the origin of land plants becomes more feasible and tractable. In this review, we integrate recent phylogenetic developments on the origin of land plants, discuss the limitations in the phylogenomics era, and provide potential directions for further research on the land plants origin.
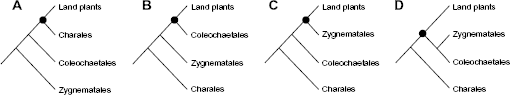
The four hypotheses for the origin of land plants. Topology shown in (A) are supported by morphological characters, 8 and the topologies shown in (B), (C) and (D) are the widely accepted hypotheses by molecular evidences. Topology (C) is currently the best hypothesis regarding the origin of land plants, though topology (D) cannot be excluded.
The Phylogenetic Progresses of Land Plants Origin
Next-generation sequencing techniques have changed the prospects for molecular evolution, and it is feasible to obtain more data at a reasonable cost. In the field of phylogenomics, which is the use of genomic data to establish and clarify evolutionary relationships, more data indeed are essential to accurately estimate phylogenetic trees (eg, reducing sampling error by increasing the amount of information; including new taxa that beak up long branches). However, it is certainly to be expected that deeper divergences will become increasingly difficult to address as we go further back in time, because the Markov models we use for sequence evolution are expected to saturate and lose some information at the most ancient divergences. 6 At shorter times, there are other potentially misleading processes happening with real populations, and a possible ancient rapid radiation at the time of terrestrial colonization by the descendants of Charophyte algae 7 could be a major factor impeding the accurate inference on the origin of land plants.
Charales, perhaps the most developmentally complex green algae, were initially suggested in an earlier period as a sister group of land plants 8 (Fig. 1A), and the early molecular phylogenetic analyses using four (two plastid, one mitochon-drial, and one nuclear) or six (four plastid, one mitochondrial, and one nuclear) genes uncovered this topology.9,10 This hypothesis was an appealing result, in that Charales appeared to have similar morphologies and growth patterns to land plants, and it supported an evolutionary scenario toward increasing cellular complexity. However, the Charales are macrophytic and coenocytic algae with multiple nuclei in large cells. 11 In contrast, Coleochaetales and Zygnematales are true multicellular algae (with plasmodesmata) that have separate cells, each with a single nucleus. In this cytological sense, Coleochaetales or Zygnematales may represent more appropriate sister groups to land plants, based on the transition from unicellular to multicellular organization.
Indeed, the genome-scale molecular data consistently reject the Charales as sister to land plants and support alternative Charophyte groups. Previous phylogenomic analyses of chloroplast genomes have yielded topology with Coleochaetales as sister to land plants12,13 (Fig. 1B), but the taxon sampling of Charophyte algae from these analyses was limited, possibly resulting in a less reliable topology. In addition, if evolutionary models do not describe the biological properties of the data, then tree building can be incorrect.14,15 Worst of all perhaps, while the use of more data could reduce sampling errors, it simultaneously makes systematic errors more apparent. Thus, not all phylogenetic problems can be easily resolved with genome-scale analyses, and more attention must be given to systematic errors when large datasets are used for phylogenetic inference.
Considering both sampling and systematic errors in genome-scale data, Zhong et al. 16 reported three new chloro-plast genomes from Charophyte algae and used a site-pattern sorting method 17 as well as site- and time-heterogeneous models18–20 to reduce both classes of errors and address the branching order among Charophyte algae and land plants. The chloroplast phylogenomic results strongly rule out earlier hypotheses placing Charales or Coleochaetales as sister group to land plants. Instead, Zygnematales alone (Fig. 1C), or a clade consisting of Zygnematales and Coleochaetales (Fig. 1D), are more likely the closest living relatives of land plants. Furthermore, this analysis indicated that more realistic models have a better fit to the data with more confidence and better infer the origin of land plants. Cox et al. 21 also supported the Zygnematales closest to land plants by reducing the compositional bias in chloroplast-genome data. Because of the highly variable structure of algae mitochondria, there are few studies investigating the origin of land plants using mitochondrial genomes. Turmel et al. 22 analyzed 40 mitochondrial protein-coding genes from Charophyte algae, but did not clearly resolve the relationship among the Zygnematales, Coleochaetales, Charales, and land plants.
Recently, the multilocus nuclear data have been commonly used to infer the origin of land plants. Phylogenomic analyses of a large number of nuclear genes have supported topologies with either Zygnematales23,24 or the branch subtending Zygnematales and Coleochaetales25,26 as closest to land plants. However, sparse taxon sampling of Charophyte algae (6 taxa 23 , 8 taxa 24 , and 10 taxa25,26) from these nuclear genome analyses cannot yet unambiguously provide the accurate phylogenetic topology. To increase the taxon sampling, Wickett et al. 27 applied RNA-Seq technology to sequence 92 transcriptomes of green plants including 18 Charophyte algae and found high support for a sister relationship between Zygnematales and land plants.
The Limitations of Genomic Data on Resolving Land Plant Origins
The large nuclear genomic data have been recently used to investigate land plant origins,23–25 but there is considerable variation (relatively low probabilities) between gene trees from different nuclear genes. The concatenation method combines different genes into a single “supergene” tree that is then considered to be equivalent to the species tree. This method was suggested to give more accurate trees than a consensus approach that summarizes congruence among individual gene trees. 28 The assumption of the concatenation method is that it assumes all genes have the same (or at least similar) gene trees,29,30 but it has become clear that individual gene trees appear to conflict with one another and gene tree heterogeneity is ubiquitous.31,32 Thus, the concatenation method may yield misleading inferences of species relationships in the presence of a high level of gene tree heterogeneity.33,34 If selecting the genes with strong phylogenetic signals (high average internode support), concatenation method may still accurately reconstruct the specie tree. 35
High gene tree heterogeneity from nuclear genes has been a significant issue in phylogenomics.31,36 There are many reasons for gene tree heterogeneity and gene trees versus species trees conflict, including horizontal gene transfer, natural selection, and incomplete lineage sorting (ILS) (Fig. 2).
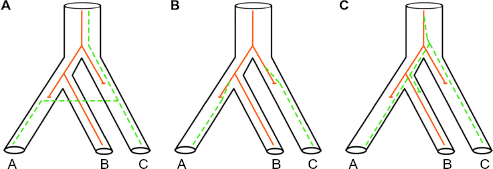
Three major biological mechanisms that can mislead phylogenetic inference – shown by the two genes (solid and dash lines respectively) of A, B, and C species not agreeing with the underlying species tree, which is ((A, B), C). (A) Horizontal gene transfer, introgression, and hybridization (all have similar consequences for the genes). (B) Natural selection (the same nonneutral mutation occurred on different lineages). (C) Incomplete lineage sorting (under lineage sorting, we expect variation of alleles in a population, but this will eventually lead to fixation of one allele).
Horizontal gene transfer (Fig. 2A): It is well accepted within evolutionary studies that there is a continuum from individuals, populations, races, varieties, sibling species, species, species complexes, subgenera, genera, etc. Along this continuum we expect introgression and hybridization to be quite normal, even if these two processes decrease at deeper divergences.
Natural selection (Fig. 2B): It has been generally assumed that most, if not all, mutations were “neutral” and that genetic drift was the dominant effect. In practice, we know very little about the factors of natural selection that might be operating in related lineages. If the mutational process is “random” (and is not related to any needs of the organism) and is occurring all the time, then there is no surprise if related lineages independently happen upon similar mutations that are advantageous.
Incomplete lineage sorting (Fig. 2C): It takes time for two variants in a population to coalesce, especially for larger populations. The failure of two or more lineages in a population to coalesce leads to the possibility that at least one of the lineages coalesces first with a lineage from a less-closely related population. 36 This factor is currently best studied and modeled as to why gene trees are distinct. The probability of inferring the wrong species tree due to ILS has been calculated theoretically for four individual species, 37 and later Pamilo and Nei 38 confirmed that ILS is a general case and proposed that adding more gene sequences will still provide the correct relationship.
In terms of investigating the origin of land plants, an ancient rapid radiation can lead to short internal and long external branches, which can increase the potential for both ILS and gene tree heterogeneity. The multispecies coalescent model is designed to approximate variation in a species tree topology derived from ILS, and it chooses ancestors from the population backward through time for multiple sequences but places some constraints on how recently the coalescences occur. Because gene trees are allowed to vary in the multi-species coalescent model, coalescent methods can consistently estimate species trees in spite of the presence of heterogeneous gene trees.39–41 Using a data set of 289 nuclear genes from 32 green plant taxa, Zhong et al. 42 applied the multispecies coalescent model for the first time to revisit the origin of land plants. In this study, the coalescent method across different subsets of data consistently suggested that the ancestors of Zygnematales are the closest relatives of land plants (Fig. 1C). In contrast, concatenation methods yield misleading inferences of species relationships in the presence of a high level of gene tree heterogeneity for the origin of land plants and support inconsistent relationships across different subsets. This analysis also shows that the multispecies coalescent model could greatly accommodate gene tree heterogeneity in deep-level phylogenies. Later, Wickett et al. 27 used similar coalescent methods with increasingly larger number of taxa and arrived at the same results. Thus, Figure 1C appears the best estimate for the origin of land plants – the Zygnematales are the closest group to land plants.
Future Perspectives
In molecular phylogenomics, Markov models are used to describe substitutions among DNA or protein sequences, and therefore to reconstruct phylogenetic trees and understand evolutionary events. When selecting the “best” model for specific data, there is always a balance between the oversimplified and overfitted models. Oversimplified models often describe the evolutionary property with only a few parameters and have the same model for all sites, possibly leading to biased conclusions. In contrast, evolutionary models that use too many parameters may have all sites to vary consistently in their rates and substitution types and overfit the data resulting in errors for estimating a large number of parameters. So it is important to evaluate whether the data can be adequately explained by evolutionary models and to identify the misfitting parts in the data.
We anticipate that a goodness-of-fit test between models and data will become a standard step in phylogenomic analyses. Similarly, we suggest that the use of more complex (well-fitted) models that incorporate heterogeneity of the substitution process will significantly improve the accuracy of phylogenetic inference. Further, with the increase of genomic data, gene tree versus species tree incongruence is becoming even more obvious, implying that biological factors may lead to “incorrect” gene trees and blur the treelike relationships. ILS appears to be the main biological mechanism resulting in gene tree heterogeneity in empirical data sets, and the mul-tispecies coalescent model should be considered as the useful tool to efficiently accommodate gene tree variation.
In general, there has been little theoretical work on the ability of methods to recover deeper divergences (eg, origin of land plants), although we cannot say that it is impossible to recover very deep phylogeny accurately, neither has it been shown that we can. In the future, we need to better understand deeper and deeper phylogeny beyond the limit of Markov models that were applied to primary sequences. We are now living in very exciting times, and the power of phylogenomics can be combined and integrated with many other aspects of biology to be able to study a wide range of questions. This has started that the origin of land plants is likely the ancestors of Zygnematales. It appears to be the single-nucleus “multicellular” lineage of green algae (rather than the “coenocytic” lineage of the Charales) that led to the “multi-cellular” land plants. Most of the Charophyte algae have motile sperms, but the current members of Zygnematales do not have motile sperms, which is assumed to be a derived feature within them. This scenario implies that there is an independent loss of motile sperms that occurred in the sister lineage of land plants. 43
We indeed need additional genome-scale data from some lineages of Charophyte algae, especially breaking up some long branches. Given that congruence of results from multiple and independent lines of evidence is a key approach for the validation of phylogenetic estimation, it is also desirable to investigate which topologies are supported with indels, gene order, and retrotransposon data. We can also include cytologi-cal features on the optimal tree with sequence data and study the evolution of the cell structure of Charophyte algae.
Author Contributions
Conceived and designed the experiments: BZ. Analyzed the data: BZ, LS, DP. Wrote the first draft of the manuscript: BZ. Contributed to the writing of the manuscript: BZ, LS, DP. Agree with manuscript results and conclusions: BZ, LS, DP. Jointly developed the structure and arguments for the paper: BZ, LS, DP. Made critical revisions and approved final version: BZ, LS, DP. All authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgments
The authors thank six anonymous reviewers for constructive criticisms on the manuscript.