
Abstract
The question how many genes are needed to resolve phylogenetic incongruence has been investigated at various taxonomic levels, yet few studies have investigated the minimum required numbers of selected genes based on single-gene tree performance at the genus level or lower. We conducted resampling analyses by compiling transcriptome-based single-copy nuclear gene sequences of 11 species of Primulina (Gesneriaceae) to investigate the minimum numbers of both random and selected genes needed to resolve the phylogeny. Only 8 of the 26 selected genes were sufficient for full resolution, while 175 genes were needed if all 830 random genes were used. Our results provided a baseline for future sampling strategies of gene numbers in molecular phylogenetic studies of speciose taxa. The gene selection strategies based on single-gene tree performance are strongly recommended in phylogenic analyses.
Introduction
Understanding the phylogenetic relationships among living organisms is fundamental to any comparative research in biology. 1 In the era of phylogenomics, phylogenetic incongruence has been widely documented in phylogeny construction with a large amount of available low-copy genes and is often ascribed to stochastic error, systematic error, and/or biological factors.2–5 To overcome phylogenetic incongruence, a practical question is how many genes should be used to generate a robust phylogenetic hypothesis. 6 This question has been investigated at various taxonomic levels.7–13 For example, one notable resampling analysis showed that a minimum of 20 concatenated genes were required to provide 95% bootstrap support for all nodes by compiling a data set from 106 random orthologous genes for seven Saccharomyces species and one out-group species. 7 In a recent study, among 59 carefully selected low-copy nuclear genes based on single-gene tree performance, fewer than 50 were enough to solve the deep angiosperm phylogeny. 13 Nevertheless, few studies have investigated the minimum required numbers of selected genes based on single-gene tree performance at the genus level or lower.
Primulina, one of the largest genera of the Old World Gesneriaceae, is a monophyletic group comprising more than 140 species widely distributed throughout southern China and adjacent countries in Southeast Asia, which is a biodiversity hot spot of the World.14,15 Nevertheless, interbreeding through artificial experiments can still succeed among many Primulina species pairs, 16 suggesting that the genus has undergone recent rapid speciation or population differentiation. Although the entire Primulina phylogeny is far from resolved to date, a rough genus framework has been presented with sequences of four loci from 104 Primulina species, 17 and the phylogeny of 11 representative Primulina species has been fully resolved based on 834 putative single-copy nuclear genes identified from transcriptome data. 18 The high species richness and endemism, and low interspecific genetic isolation, together with comparative transcriptomic resources, make Primulina an ideal model system to study the gene numbers needed to obtain a robust phylogeny. In this study, we conducted resampling analyses by compiling the previously published transcriptome-based single-copy nuclear gene sequences of 11 representative Primulina species 18 to determine the minimum numbers of both random and selected genes needed to resolve the phylogeny and to provide a baseline for future sampling strategies of gene numbers in molecular phylogenetic studies of speciose taxa.
Materials and Methods
The aligned sequence data for 830 putative single-copy nuclear genes (Supplementary Table 1) from 11 Primulina species 18 were used for analysis, while the other four loci were discarded due to poor alignment performance. To test the utility of individual genes, all single-gene trees were constructed with 1,000 bootstrap replicates based on nucleotide sequences, using the maximum-likelihood (ML) method implemented in PhyML v3.0. 19 Primulina swinglei was chosen as out-group according to the Primulina phylogeny. 17 The nucleotide substitution model was specified as GTR+I+γ and the branching-swapping method was set to subtree pruning and regrafting. To consider the limitations of the concatenation approach for phylogenetic inference, 20 the average rank of coalescences (STAR) method 21 implemented in the species tree analysis web server STRAW 22 was conducted based on the 830 single-gene trees to infer the species tree of the 11 Primulina species. Those genes based on which the topologies of the 11 Primulina species were congruent with the species tree were screened as good genes. To visualize the phylogenetic incongruence among different topologies, SplitsTree v4.13.1 23 was used to infer the consensus network from 830 source singlegene trees with a set of threshold values (0.01, 0.05, 0.1, 0.15, 0.2, and 0.25). Different threshold values were set to control the visual complexity of resulting networks by using only the splits that occurred in more than a given proportion of all trees.
Summary of the percentages of correct trees at a bootstrap threshold of 95% or 75% (in parentheses) for all 11 Primulina species, the triplet, and other species, through random resampling by gene or site from the original 830-gene (631,686-nucleotide) data set.
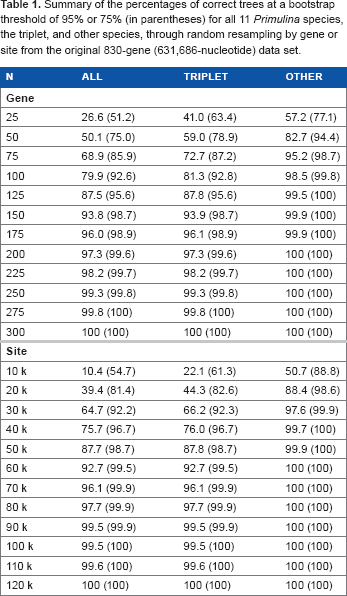
To investigate the effect of the number of sampled genes or nucleotides on the probability of the inferred species tree, random resampling without replacement from the original 830-gene (631,686-nucleotide) data set was performed with a custom java script. The number of sampled genes started from 25 with increments of 25, until the percentage of correct trees (see below) reached 100. Similarly, resampling by site started from 10 kb with increments of 10 kb. For each sample, 1,000 replicates were generated and the ML gene trees were inferred by RAxML v8 24 with the model GTR+γ and 100 fast bootstrap replicates. Those trees with the same topology as the species tree and all ML bootstrap values larger than a threshold (95% or 75%) were counted as correct trees. Gene resampling among the selected good genes was also conducted and started from three with increments of one.
Results
The inferred STAR species tree (Fig. 1) showed the same topology as the previously published Primulina topology based on concatenated data 18 and all bootstrap values were 100. Only 26 of the 830 single-gene trees exhibited the same topology as Figure 1 (Supplementary Tables 1 and 2) and there were 25 topologies supported by more than three genes (Supplementary Table 3), indicative of extensive single-tree phylogeny incongruence, which was also strongly supported by the consensus network constructed from 830 source single-gene trees (Fig. 2). Network complexity decreased as increasing threshold values and ended up with a topology identical to that shown in Figure 1 when the threshold reached 0.25, in which all boxes were collapsed. Among the 11 species, the triplet of Primulina fimbrisepala, Primulina dryas, and Primulina villosissima was the most difficult clade to resolve. Of the 830 single-gene trees, 404 (48.7%) supported the correct sister relationship between P. fimbrisepala and P. villosissima, while those supporting the alternative sister relationship between P. fimbrisepala and P. dryas or between P. dryas and P. villosissima numbered 206 (24.8%) and 220 (26.5%), respectively (Supplementary Table 1), suggesting extremely rapid triplet speciation. These results further support the previously published Primulina phylogeny 18 and highlight the phylogenetic incongruence of the triplet of P. fimbrisepala, P. dryas and P. villosissima.
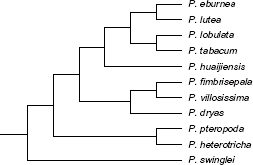
Species tree of the 11 Primulina species inferred from the 830 single-gene trees with the average rank of coalescences (STAR) method.
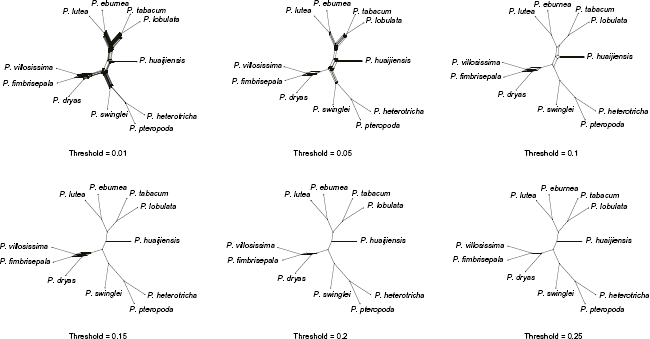
Consensus network of the 11 Primulina species constructed from 830 single-gene trees at six different threshold values.
Summary of the percentages of correct trees at a bootstrap threshold of 95% or 75% (in parentheses) for all 11 Primulina species, the triplet and other species, through random resampling by gene from the selected 26-gene data set.
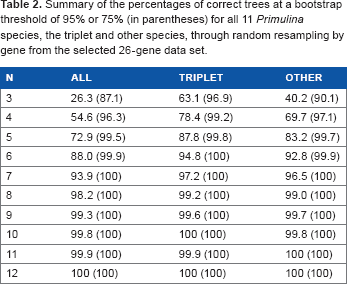
The resolution performance through resampling by gene or site demonstrated that the probability of getting a solid topology steadily increased with the number of genes or sites sampled (Table 1). Using 95% correct trees with a 95% bootstrap threshold in 1,000 replicates as the criterion, about 175 genes were needed to resolve all 11 species, and about 75 genes were needed when ignoring the triplet complexity. When resampling by nucleotide site, about 70 kb of nucleotides yielded sufficient resolution, equivalent to 92 sampled genes given the average length of 761 bp per gene, and it took about 30 kb (about 39 genes) for the other clades except the triplet. Nevertheless, when resampling by gene among the 26 selected genes (Supplementary Table 2), only 8 genes were needed to resolve all the clades. As expected, all the counts decreased somewhat when using a bootstrap threshold of 75% rather than 95%. Under a bootstrap threshold of 75%, 4 selected genes were sufficient for full resolution, while 125 genes were needed if all 830 random genes were used.
Discussion
As proposed previouly,6–8,13 the inferred minimum gene number needed to resolve the phylogeny should be related to bootstrap threshold values to determine a correct tree, topological complexity of sampled species, and selection of source gene sets. The differences caused by the two bootstrap threshold values, 95% and 75%, were substantial in this study (Tables 1 and 2). Similar to our results, another study found that the gene number dropped from 20 to 8 when decreasing the bootstrap threshold value in a set of eight yeast species. 7 Nevertheless, the stricter criterion with a 95% threshold will provide a better estimate of the minimum gene number.
As expected, the required gene numbers for the Primulina phylogeny fell sharply from 175 to 75 when ignoring the most difficult triplet (Table 1). Similarly, the suggested gene number of 20 based on the eight yeast species set 7 was unnecessary for fewer taxa. 6 In fact, it is difficult to judge whether the test species set in this study is representative for other work, especially when focusing on topological complexity characterized by the number of sampled species (11) and the pairwise interspecies genetic distances (Ks: 0.027–0.064, Supplementary Table 4). Nevertheless, the required gene number of 75 when ignoring the triplet complexity in this study is still unrealistic for routine laboratory work with large-scale species sampling. Although unlinked nucleotide sites could provide more efficient phylogenetic performance than genes in terms of better data independence, as shown in this study (Table 1) and the previous studies,7,11 most experimental data in molecular phylogenetics are still in the form of amplified gene fragment sequences.
To minimize the effect of hidden paralogs and to identify the most probable orthologs, one optimized sampling strategy is to select genes by checking single-gene trees, rather than to use all available random genes as in most previous studies. 13 In this study, we followed such a gene selection method based on single-gene tree performance 13 and found that only 8 of 26 selected genes were sufficient to resolve all 11 Primulina species, while 175 genes were needed if all 830 random genes were used (Tables 1 and 2). Compared with the two factors discussed above, selection of source gene sets has greater effect on the inferred minimum gene number. Based on our results, we strongly recommend the gene selection strategies based on single-gene tree performance of a few representative species with known or well-resolved phylogenies, and the subsequent use of the selected genes for phylogenetic reconstruction with larger scale species sampling.
Author Contributions
Conceived and designed the experiments: BA, MK. Analyzed the data: BA. Wrote the first draft of the manuscript: BA. Contributed to the writing of the manuscript: BA, MK. Agree with manuscript results and conclusions: BA, MK. Jointly developed the structure and arguments for the paper: BA, MK. Made critical revisions and approved final version: BA, MK. Both authors reviewed and approved of the final manuscript.
Supplementary Materials
Supplementary Table 1
List of alignment lengths, proportions of variable sites, single-gene topologies, and function associations of the 830 genes.
Supplementary table 2
List of alignment lengths, proportions of variable sites, and function associations of the selected 26 genes.
Supplementary Table 3
List of 25 topologies supported by more than three genes.
Supplementary Table 4
List of the pairwise interspecies divergence (Ks) values among the 11 Primulina species, estimated by Ai et al. 18
Footnotes
Acknowledgments
We thank Xinhui Zou and Fumin Zhang for their help in the data analysis.