
Abstract
As a result of the development of rapid and efficient sequencing technologies, complete sequences of numerous mitochondrial genomes are now available. Mitochondrial genomes have been widely used to evaluate relationships between species in several fields, including evolutionary and population genetics, as well as in forensic identification and in the study of mitochondrial diseases in humans. However, the creation of mitochondrial genomes is extremely time consuming. In this paper, we present a new tool, MITOSCISSOR, which is a rapid method for parsing and formatting dozens of complete mitochondrial genome sequences. With the aid of MITOSCISSOR, complete mitochondrial genome sequences of 103 species from
Introduction
Mitochondria are intracellular organelles that have their own DNA, distinct from the nuclear genomes of the cells in which they reside. Throughout evolution, mitochondrial genomes (mitogenomes) have accumulated many small genetic changes that differ between species, and which allow biologists to infer the phylogenetic relationships between species, and even bet ween subpopulations within a species.
The analysis of mitochondrial DNA (mtDNA) has contributed enormously to our understanding of evolution. The rapid rate of mtDNA sequence divergence compared to that of the nuclear genome (due to a relatively high nucleotide substitution rate and inefficient DNA repair in the mitochondria) allows the discrimination of recently diverged lineages.
1
Mitochondrial genes that are frequently used for these analyses include cytochrome b (
In addition to the insights that mtDNA can provide on the differences between closely related organisms, the sequencing of mitogenomes is much more cost effective than the analysis of entire nuclear genomes. Thus, the popularity of the complete mitogenome as a phylogenomic marker is increasing.6–9
Comparative analysis of mitochondrial genomes that have been sequenced and annotated thus far has resolved many controversial phylogenetic issues, including inconsistencies regarding both lower level and higher level relationships. 9 However, creation of these DNA datasets is time consuming when these mitogenomic sequences must be manually parsed. For example, the conventional method for manually constructing such mitogenome matrices involves the following steps: (1) Fasta sequences of all the selected mitochondrial genomes are downloaded from GenBank or other public databases. (2) The sequence of every gene in each mitochondrial genome is determined and then saved as an individual fasta file. A large number of files are generated during this process, and the compilation is cumbersome. For example, the mitogenomic analysis for 100 vertebrate species will generate 3,700 files, since there is on average 37 genes in one vertebrate mitochondrial genome. (3) All of the sequences are then arranged in the same orientation using sequence-editing software such as BioEdit or DNASTAR, and the stop codons are removed from protein-encoding genes. (4) Finally, the protein-coding genes, rRNA genes, and tRNA genes are concatenated head to tail to form a single supergene for the evolutionary analysis. Obviously, parsing mitogenomic sequences is very time consuming, and the workload to analyze so many genes is enormous without the use of processing tools.
Here we present a useful Perl script named MITOSCISSOR, which automatically executes the parsing work for mitogenomic data. As an example, we applied MITOSCISSOR for creating mitogenomic data sets for 103 Tetraodonti-form fishes. Phylogenetic relationships within this order were also inferred, providing new insights into the relationships within this order. It should be noted here that MITOSCISSOR is limited to datasets containing mitogenomes with the same overall gene order. This problem will be solved with the growth of data. The development of MITOSCISSOR will increase the ease of investigating positive selection in marine fish (and other organisms), by its ability to quickly parse out genic regions of interest.
Materials and Methods
Taxonomic sampling
Mitogenome sequences of 103 Tetraodontiform fishes and 9 fish species from other orders were employed in the present study. The corresponding GenBank accession numbers (Supplementary File 1) were searched in the MitoZoa database (http://srv00.ibbe.cnr.it/mitozoa/), and the complete L-strand nucleotide sequences from these mitogenomes (.gb file) were downloaded using the NCBI batchentrez tool (http://www.ncbi.nlm.nih.gov/sites/batchentrez).
Mitoscissor Implementation
MITOSCISSOR was written in the Perl language. Activeperl (http://www.perl.org/get.html) should be installed before using MITOSCISSOR in Windows OS.
The MITOSCISSOR software and all the GenBank files should be saved together in a single folder. Once these steps are performed, the command line interpreter cmd.exe would be started and automatically linked to the working directory. Users then doubleclick “MitoScissor.exe”, and the window interface would be presented, as shown in Figure 1. Processing could be easily done according to the instructions.

Window OS interface of MITOSCISSOR.
In the Start Menu (operation system such as Windows OS) search box, users could type in “command prompt” or “cmd”, then carry out the following operations >dir (“dir” command shows there are three files in the working directory). Subsequently, the software could be executed by typing a simple command line, “perl MitoScissor.pl sequence.gb outputfile”. Note that “printall” is an optional parameter used in cases where the Genbank file does not meet recognition models and the operator wants to output all sequences that can be identified.
In this procedure, the user should choose an appropriate module model. The organization of the mitochondrial genome is generally conserved among vertebrates, but some variations between higher taxonomic levels have been found.10,11 Therefore, we designed different recognition models with regard to specific taxa. Here, we chose the module of Fish.
In the current version of MITOSCISSOR, a Windows OS interface (shown in Fig. 1) was designed to provide greater operational convenience for users.
With the help of MITOSCISSOR, mitogenomic datasets were created in three simple steps. (1) GenBank accession numbers for complete mitochondrial genomes were searched and listed. (2) The corresponding GenBank files (hereafter called inputfile.gb) containing the complete mitochondrial genome sequences were obtained from public databases. (3) The downloaded GenBank files and MITOSCISSOR were placed in the same directory. Once these steps have been performed, the cmd.exe (a command in Windows OS) is opened, and MITOSCISSOR is run by typing the simple command, “perl MitoScissor.pl sequence.gb outputfile” (the process is shown in Fig. 2).

Flow-process diagram in the software MITOSCISSOR.
Tests of positive selection
The PAML package12 was used to estimate parameters in substitution models and to reveal whether positive selection was occurring. In PAML, there is a basal ratio model that assumes a single average ratio of nonsynonymous versus synonymous substitution rates (CO =
Sequence data and phylogenetic analyses
Sequences were then subjected to multiple alignments using MAFFT. 14 Gblocks was adopted to trim both the ambiguous alignments and highly diverged regions of the alignment via the “more stringent selection” setting. 15 Pairwise nucleotide differences in evolutionary distance were plotted in order to examine the pattern of sequence substitution with DAMBE. 16 Modeltest 3.06 17 was used to determine the most appropriate nucleotide substitution models with Akaike information criterion (AIC). 18 Analyses of phylogenetic relationships were conducted in a Bayesian framework using the software package MrBayes3.1.2. 19 Bayesian analyses were run five times for 50 million generations to ensure the convergence of Markov chain Monte Carlo (MCMC) chains. MCMC chains were run until convergence was reached, as determined by the standard deviation of the split frequencies dropping below 0.01.
Results and Discussion
It took less than one minute for MITOSCISSOR to assemble the 103 Tetraodontiform mitochondrial genomes that we had selected and to split these genomes into four partitions, each with their own fasta file. The partitions were as follows: protein genes, tRNA genes, rRNA genes, and individual genes. The corresponding fastafiles were output_proteingene.fa, output_rRNA.fa, output_tRNA.fa, and individual gene.fa. The results of this analysis are shown in Supplementary Files 2–5. The size of the Tetraodontiform mitochondrial genomes we examined ranged from 16,418 bp to 16,649 bp (shown in Supplementary File 6). The software conserves codon position order in the case of protein-coding genes that have an incomplete start or stop codon for analyses requiring codon-partitioning by inserting gaps.
The accuracy of molecular phylogeny primarily hinges on the selection of unsaturated genes.20,21 Iss and Iss.c values were calculated with the aid of the program EVOLVER in the PAML package.
12
The index score (Iss = 0.4361) was significantly lower than the critical score (Iss.c = 0.7529) (
All nine Tetraodontiform families were clearly resolved as monophyletic with high support and were clearly revealed in Bayesian analysis (GTR+I+G model), and sister-group relationships were established (the minimum posterior probability value = 0.91) (Fig. 3). The topological inference from phylogenetic relationships in this study showed that subfamily Tetraodontinae is paraphyletic, which is in agreement with phylogenetic analyses based on comparative skeletal anatomy.22,23
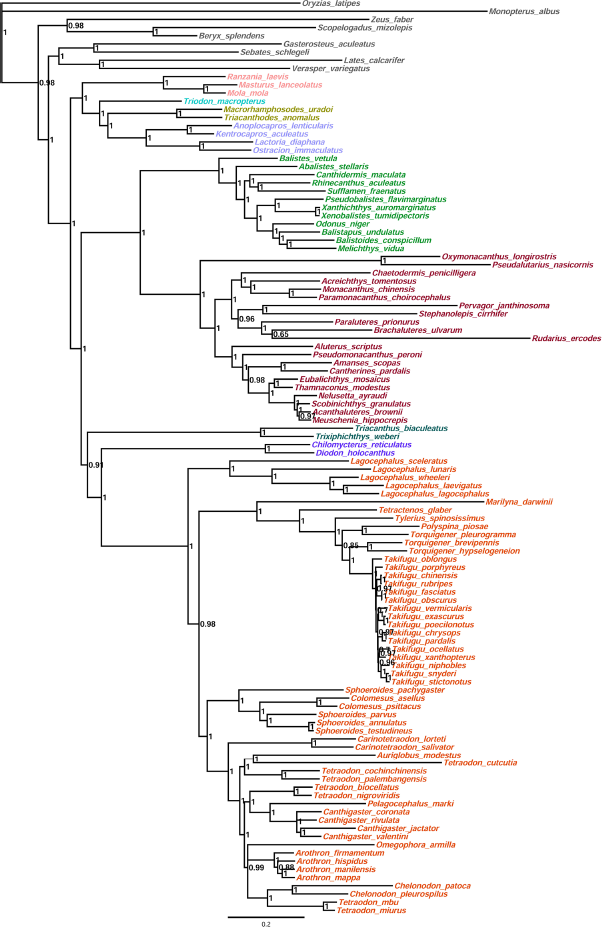
Bayesian phylogenetic tree of the order
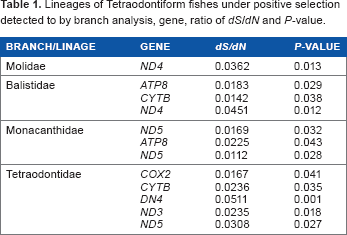
Detecting positive selection is generally difficult because the signal may be swamped by the ubiquitous negative selection, but the proposition that mitochondrial genes have been subjected to non-neutral evolution has been gaining support in the last decades.4,20 Many studies have uncovered some incidences of positive selection in several genes in the fish. Examples include the cytochrome c oxidase subunit (

Bar chart showing the proportion of codons under significant positive selection in each gene for Tetraodontiform fishes.
Lineages of Tetraodontiform fishes under positive selection detected to by branch analysis, gene, ratio of
Conclusions
We have used MITOSCISSOR to analyze 103 Tetraodontiform mitogenome sequences and established the first robust molecular phylogeny with clear resolution for interfamilial relationships in the order
We compiled a new software, MITOSCISSOR, for parsing and formatting complete mitochondrial genome sequences. MITOSCISSOR was much more reliable and efficient compared to manual editing.
We established the first robust molecular phylogeny with clear resolution for Tetraodontiform fishes.
Positive selection was detected in nine mitochondrial genes or Tetraodontiform fishes.
Author Contributions
Designed the software MITOSCISSOR: ZS, YZC, JBZ. Phylogentic analysis: ZS, YZC. Prepared the manuscript: JBZ, ZS. All authors reviewed and approved of the final version of the manuscript.
Supplementary Material
Supplementary File 1
Complete mitochondrial genome sequences used in this study.
Supplementary File 2
Sequences of protein genes for 103 Tetraodontiform fishes.
Supplementary File 3
Sequences of tRNA genes for 103 Tetraodontiform fishes.
Supplementary File 4
Sequences of rRNA genes for 103 Tetraodontiform fishes.
Supplementary File 5
Split individual genes for 103 mitochondrial genomes of Tetraodontiform fishes.
Supplementary File 6
General information about mitochondrial genomes employed in this paper.
Footnotes
Acknowledgments
Thanks for the academic editor and reviewers for their comments, which were very helpful in improving the manuscript.