
Abstract
Optimizing microalgal biofuel production using metabolic engineering tools requires an in-depth understanding of the structure-function relationship of genes involved in lipid biosynthetic pathway. In the present study, genome-wide identification and characterization of 398 putative genes involved in lipid biosynthesis in Arabidopsis thaliana Chlamydomonas reinhardtii, Volvox carteri, Ostreococcus lucimarinus, Ostreococcus tauri and Cyanidioschyzon merolae was undertaken on the basis of their conserved motif/domain organization and phylogenetic profile. The results indicated that the core lipid metabolic pathways in all the species are carried out by a comparable number of orthologous proteins. Although the fundamental gene organizations were observed to be invariantly conserved between microalgae and Arabidopsis genome, with increased order of genome complexity there seems to be an association with more number of genes involved in triacylglycerol (TAG) biosynthesis and catabolism. Further, phylogenomic analysis of the genes provided insights into the molecular evolution of lipid biosynthetic pathway in microalgae and confirm the close evolutionary proximity between the Streptophyte and Chlorophyte lineages. Together, these studies will improve our understanding of the global lipid metabolic pathway and contribute to the engineering of regulatory networks of algal strains for higher accumulation of oil.
Introduction
Growing levels of atmospheric pollution, mounting energy demand, and the incessant rise in crude oil prices are some of the issues which have in recent times driven global efforts in biofuel research. Currently, commercial-scale biofuels are sourced primarily from a variety bioenergy crops that include sugarcane (Saccharum officinarum), sugar beet (Beta vulgaris), switch grass (Panicum virgatum), soybean (Glycine max), canola (Brassica napus) and sunflower (Helianthus annus). 1 Although the environmental benefits of biofuels as compared to fossil fuels are well established, concerns are being raised about their long-term sustainability, especially against the backdrop of diversion of arable land for biofuel-based cropping systems and their corresponding adverse impact on the global food supply chain. 2 In consequence, algae-based biofuels are increasingly gaining the attention of researchers due to their rapid growth rate coupled with high carbon dioxide uptake, high lipid content and comparatively low, marginal land usage rates. 3
Notwithstanding the many advantages of biofuels and their technical feasibility, the commercial viability of the algal biofuel process is still an area of concern requiring better strain development and improved post-harvest process engineering. 4 The major challenge is to achieve accumulation of improved lipid profiles with concomitant reduction in energy inputs in order to minimize the cost of production. 2 The enhancement of lipid production in microalgal cells under controlled stress conditions and engineering metabolic pathways are promising strategies to obtain large amounts of standard biofuel for industry. Despite positive experimental reports on enhanced microalgal lipid accumulation under physiological or nutritional stress regimes, many contrasting studies have indicated a concomitant reduction in overall biomass yield under such conditions. 5 In this context, harnessing the potential of genome-scale metabolic engineering has been suggested as a promising area of research to boost oil production in microalgal strains, including modification of algal lipid profile for improved biofuel properties.6,7
Over the past few years various studies have been carried out concerning alteration of fatty acid composition in plants through genetic engineering approaches, along with the development and deployment of a number of plant lipid-related genomics databases.8–11 Comparative genomics analyses using bioinformatics tools have also been performed recently to identify genes involved in lipid biosynthesis in various oleaginous plants. For example, a total of 1003 maize lipid-related genes were cloned and annotated by Lin et al, 12 while Sharma and Chauhan 13 identified a total of 261 lipid genes from the genome of Arabidopsis, Brassica, soybean and castor. Complete or near complete genome sequences have been reported for several algae. 6 Yet, lack of adequate knowledge regarding the structure-function of lipid biogenesis genes in an evolutionary context is a major impediment in engineering metabolic pathways of algae for over-production of fuel precursors. 14 Various experimental techniques like insertional mutagenesis and targeted gene disruption have been employed to analyze gene function in a few algae. However, many of these approaches are tedious, time-consuming, fiscally prohibitive and limited by a number of biological constraints. 15 As an alternative, phylogenomics is now increasingly used to gain insights into metabolic pathways at the molecular level by comparative genomics and co-evolutionary analyses of related gene. 16 Therefore the present work was designed to identify the genes involved in lipid metabolic pathway from the genomes of microalgae (including Chlamydomonas reinhardtii, Volvox carteri, Ostreococcus lucimarinus, Ostreococcus tauri and Cyanidioschyzon merolae) using sequence similarity search with Arabidopsis thaliana homologs. In addition phylogenomics protocols have been employed to study the structure-function relationship of the encoded proteins and to gain much needed insights into their phylogenetic evolution. We hope that the present study contributes to the biochemical and molecular information needed for augmentation of lipid synthesis in microalgae.
Materials and Methods
Gene retrieval and annotation
An initial set of lipid genes was obtained from the Arabidopsis thaliana lipid gene database (http://www.plantbiology.msu.edu/lipids/genesurvey/index.html) to construct a query protein set. The Arabidopsis lipid gene database is a convenient and reliable source of genes covering all the major biochemical events responsible for biosynthesis and catabolism of plant lipids. 17 Subsequently, each protein in the query dataset was used to identify homologs in microalgae by subjecting it to BLASTp 18 search with e-value inclusion threshold set to 0.001 against microalgal genome databases provided by Joint Genome Institute. These include Cyanidioschyzon merolae http://merolae.biol.s.u-tokyo.ac.jp/), Chlamydomonas reinhardtii (http://genome.jgi-psf.org/chlamy/chlamy.info.html), Volvox carteri (http://www.phytozome.net/volvox.php), Ostreococcus lucimarinus (http://genome.jgi-psf.org/Ost9901_3/Ost9901_3.home.html), Ostreococcus tauri (http://genome.jgi-psf.org/Ostta4/Ostta4.home.html). Based on multiple alignments and/or the presence of conserved motif patterns, some initial sequences “hits” were then discarded. Functional descriptions of genes or gene products were performed by annotation of Cluster of Orthologous groups (COGs) using KOGnitor program, 19 the latter being a widely used tool in the field of computational genomics for detecting candidate set of orthologs in prokaryotes and eukaryotes. 19 In addition, assignment of Gene Ontology (GO) terms describing biological processes and molecular function was annotated by the GO browser and annotation tool AmiGO. 20 The Gene Ontology is currently the pre-eminent approach for functional annotation of homologous genes and protein sequences in multiple organisms. 20
Metabolic pathway study
Metabolic pathways were subsequently analyzed using the KEGG pathway database, 21 an extensively employed biochemical pathway database to analyze lipid pathways in diverse organisms. 22 To enrich the pathway annotation, sequences were submitted to the KEGG Automatic Annotation Server (KAAS) to identify the orthologous gene groups. 23 KAAS annotates every submitted sequence with a KEGG ortholog (KO) identifier that allows identification of orthologous and paralogous relationships between the genes of interest. Further, a set of six reference pathway maps, namely fatty acid biosynthesis, fatty acid metabolism, fatty acid elongation, glycerolipid metabolism, glycerophospho-lipid metabolism and pathway map for biosynthesis of unsaturated fatty acids, were downloaded from the KEGG database. This dataset contains a complete biochemical description of the pathways related to the lipid metabolism observed in different organisms. They were used as templates for comprehensive examination of the lipid biosynthetic genomic repertoire of microalgae by correlating genes in the genome with gene products (enzymes), in accordance with their respective Enzyme Commission (EC) number.
Prediction of subcellular localization
Three different protein targeting prediction programs were used to determine the putative subcellular localization of the candidate proteins: TargetP, 24 ChloroP 25 and WolfPsort. 26 Each program is based on different terminology and predictions. The location assignment of TargetP is based on the presence of any of the N-terminal presequences: chloroplast transit peptides (cTP), mitochondrial targeting peptide (mTP) or secretory pathway signal peptide (SP). The ChloroP server predicts the presence of chloroplast transit peptides (cTP) in protein sequences and the location of potential cTP cleavage sites. WolfPsort is an extension of the PSORT II program for protein subcellular localization prediction. It classifies protein into more than 10 location sites, including dual localization such as proteins which shuttle between the cytosol and nucleus. The sensitivity and specificity of this program has been experimentally verified to be 70%.
Physico-chemical characterization and secondary structure prediction
Physico-chemical properties like length, molecular weight, isoelectric point (pI), total number of positive and negative residues, Instability Index, 27 Aliphatic Index 28 and Grand Average hydropathy (GRAVY) 29 were computed using the Expasy's ProtParam server. 30 GOR IV server 31 was employed for the prediction of secondary structural features like alpha helices, extended strands and random coils in terms of percentage in the protein sequences.
Calculation of the GC content
The GC content of the predicted genes was determined using Genscan web server. 32
Motif identification
Protein sequence motifs for each gene family were identified using the MEME program. 33 The analyses parameters were set as follows: number of repetitions-zero or one per sequence; maximum number of motifs—1; minimum and maximum width—6 and 50, respectively. The motif profile for each gene family is presented schematically. Domain arrangements along sequences were predicted using InterProscan 34 to determine protein homolog relationships among species.
Exon-intron structure and phylogenetic analyses
The exon-intron structural patterns of the lipid biosynthetic genes were analyzed using the gene prediction algorithm of Genscan. 32 To construct the phylogenetic tree, amino acid sequences were aligned using the ClustalX program implemented in BioEdit 35 (v 7.1.3) with default settings and then manually refined by trimming of poorly conserved N and C termini. ClustalX 36 has been demonstrated to be a user-friendly tool for providing good, biologically accurate alignments within a reasonable time limit. Many options are provided such as the realignment of selected sequences or blocks of conserved residues and the possibility of building up difficult alignments, making ClustalX an ideal tool for working interactively on alignments. 36 Subsequently, sequence alignment of genes predicted to be in similar families were used as an input file for the MEGA 4 software. 37 Phylogenetic tree was built via the neighbor-joining (NJ) method with evaluation of 1000 rounds of bootstrapping test, followed by identification of sub-tree.
Results and Discussion
Comparative genomic analyses of lipid genes in microalgal species
Interest in microalgae as a potential feedstock for biofuel production and other valuable biomaterials is rooted in the ability of microalgae to rapidly accumulate significant amounts of neutral lipids. 38 Under optimal conditions, microalgae synthesize fatty acids used primarily for esterification into polar glycerol-based membrane lipids like glycosylglycerides and phosphoglycerides, whereas under stress conditions, many microalgae tend to accumulate storage lipids called triacylglycerol (TAGs). 16 Although global fatty acid biosynthetic mechanisms are known in higher plants, 39 pathways responsible for lipid accumulation in microalgae are not well studied. Hence, in order to bridge our existing knowledge gap regarding algal lipid metabolism, comparative metabolic pathway analyses have been performed across five microalgal genomes, using homologous plant genes as reference with an objective of functional characterization of predicted genes. EC numbers, Cluster of Orthologous Groups (COGs), protein domain family and GO terms were determined for the respective candidate genes. The above in silico approach has been reviewed recently to be reliable enough for accurate function prediction of uncharacterized proteins encoded by genes in a genome. 40
In the present study, using the Arabidopsis annotation data as the BLAST input query set, a total of 398 orthologous genes present in A. thaliana, C. reinhardtii, V. carteri, O. lucimarinus, O. tauri and C. merolae genomes were identified. The above approach to identify candidate genes involved in biosynthesis and accumulation of storage oil has been successfully demonstrated in plants by Sharma and Chauhan. 13 These 398 genes clustered into 40 gene families and includes 142, 56, 59, 47, 41 and 53 genes from A. thaliana, C. reinhardtii, V. carteri, O. lucimarinus, O. tauri and C. merolae genomes, respectively (Table 1). The identified genes are involved in the synthesis of phospholipids, glycerolipid and storage lipids like TAG. We further divided the predicted genes into categories like gene-coding enzymes involved in biosynthesis and catabolism of fatty acid, TAG and membrane lipid. The comprehensive list of candidate genes along with experimental evidence of the respective enzyme action influencing lipid accumulation is presented in Table 1.41–74 Approximately 47% of the predicted gene products found in the present study were previously annotated as ‘predicted’, ‘probable’, ‘putative uncharacterized’ and ‘similar’ or ‘hypothetical’ proteins (Table 1). The annotation of these sequences has been improved and a role in lipid biosynthetic process was assigned to each of them by similarity search with homologous plant genes, annotation of Gene Ontology, and through identification of conserved domains or motifs. Furthermore, on comparison to the previous report on lipid gene identification in C. merolae genome by Sato and Moriyama, 75 the present study has identified 20 additional genes involved in lipid biosynthesis.
Table I. Candidate genes involved in lipid biosynthetic pathway of Arabidopsis thaliana, Chlamydomonas reinhardtii, Volvox carteri, Ostreococcus lucimarinus, Ostreococcus tauri and Cyanidioschyzon merolae genome.

Putative uncharacterized proteins;
predicted proteins;
probable proteins;
similar protein;
absent in KEGG pathway database;
relevant references on experimental evidences of the respective enzyme action influencing lipid accumulation.
To investigate metabolic processes responsible for the synthesis of microalgal biofuel precursors, KO identifiers were assigned to the predicted 398 genes representing 36 unique EC numbers, which were subsequently used to study metabolic pathway maps available in KEGG pathway database. KEGG is considered one of the most important bioinformatics resources for understanding higher-order functional meaning and the utilities of the organism from its genome information. It hosts information on the majority of well-known metabolic pathways, including lipid pathways for several organisms such as higher plants, bacteria and algae. Recently, it has been used successfully by Rismani-Yazdi et al 14 to identify pathways and the underlying gene responsible for production of biofuel precursors in Dunaliella tertiolecta, a potential microalgal biofuel feedstock. Using the above approach, a total of 79 lipid genes including 22 from A. thaliana, 21 from C. merolae, 10 from C. reinhardtii, 10 from O. lucimarinus and 8 each from V. carteri and O. tauri were recognized that were not earlier indexed in KEGG metabolic pathway database (Table 1).
The global synthesis pathway of TAG begins with the basic fatty acid precursors, acetyl-CoA, and continues through fatty acid biosynthesis, complex lipid assembly and saturated fatty acid modification until TAG bodies are finally formed. 76 A simplified overview of TAG biosynthetic pathway in microalgae is shown as Figure 1. Comparative analyses with the genomes of C. reinhardtii, V. carteri, O. lucimarinus, O. tauri, C. merolae and A. thaliana indicates that the majority of genes involved in lipid production are orthologous among these species. Additionally, the extensive amino acid sequence conservation (more than 60% pair-wise sequence identity) among the genes involved in lipid biosynthesis provides indications of functional equivalence between Arabidopsis and microalgal genes. Thus, the present results demonstrate that the underlying fatty acid and TAG biosynthesis process are directly analogous to those reported in higher plants. 16 It may further be noted that although algae predominantly share similar lipid biosynthetic pathways with higher plants, the present in silico analyses revealed that the sizes of the gene families responsible for lipid biosynthesis in microalgae are smaller than Arabidopsis. Certain specific pathways were also observed to be absent in microalgae, including the fatty acid biosynthesis termination mechanism by FAT homologs in C. merolae. The above computational analyses find support from the previous experimental reports on the algal lipid metabolism. 75

Schematic overview of Triacylglyceride (TAg) biosynthetic pathway in microalgae.
Furthermore, our results conclusively indicate that enzymes that are responsible for higher lipid accumulation in plants and other eukaryotes, either through over-expression or gene knockout strategies, are present not only in oleaginous algal species (C. reinhardtii) but also in other algal species, notably O. tauri and C. merolae (Fig. 2). Comparison of the number of genes in each step of lipid metabolic pathway suggests that the green algae C. reinhardtii and V. carteri have an expanded array of genes involved in TAG biosynthesis and catabolism, including fatty acid thioesterase, long chain acyl-CoA synthase, acyl-CoA oxidase, desaturase, glycerol-3-phosphate acyltransferase, and diacylglycerol acyltransferase. Additionally, the proportion of these gene copy numbers appear to be correlated with the genome complexity of the organisms under study (Fig. 2).

Number of gene homologues in the TAG biosynthetic pathway in A. thaliana, C. reinhardtii, V. carteri, O. lucimarinus, O. tauri and C. merolae.
Prediction of subcellular location
The prediction of subcellular localization of proteins is essential to elucidate the spatial organization of proteins according to their function and to refine our knowledge of cellular metabolism. 77 Thus, prediction of subcellular location provides valuable information about the function of proteins as well as the interconnectivity of biological processes. 78 In the present study, subcellular location of lipid biosynthetic proteins by tools such as TargetP, ChloroP and WolfPsort showed different locations using several unique algorithms. The objective of using more than one analytical tool was to improve the specificity of the prediction, as various studies have shown that combined results from several prediction programs are advantageous to rule out false positives and false negatives. 78 The available localization prediction tools show different strengths and no tool is clearly and globally optimal. 77 Moreover, it is known that some localizations are badly predicted by all the algorithms, especially in the case of proteins exhibiting dual targeting to plastids and mitochondria, which could be a phenomenon more common than previously thought. 79 This analyses showed that majority of the predicted proteins are located in four compartments: plastids (31%), mitochondria (26%), cytoplasmic (28%) and nucleus (6%) (Fig. S1 and Table S1). The above results are consistent with the experimental observations that de novo synthesis of fatty acids occurs primarily in the plastid and/or mitochondria. 5 About 19% of the proteins revealed the presence of both the mitochondrial target peptide and chloroplast transit peptide in the sequences. Recent reports have shown an unexpectedly high frequency of dual targeting of proteins to both the mitochondria and chloroplast, hence making it difficult to predict the correct location of these proteins within a cell.80,81 Furthermore, approximately 3% of the predicted proteins were located in more than one compartment ie, nucleus and cytoplasm, which were the same highly paired compartments as identified in Arabidopsis 82 and sugarcane 83 proteome, suggesting that there is a significant amount of interactions between these two organelles.
Hyunjong et al 84 have reported that targeting a particular enzyme to several compartments simultaneously in the same plant will augment its production when compared to its individual compartments in the same plant. Hence the predicted localization information would certainly aid in targeting the lipid biosynthetic enzymes to enhance oil accumulation in microalgae.
Physico-chemical characterization and secondary structure prediction
Various physico-chemical parameters were computed using Expasy's ProtParam tool (Fig. 3 and Table S2). Molecular weight was observed between the ranges of 1116.818–299171.0 for all lipid biosynthetic proteins in microalgae. The majority of the predicted proteins were found to have a pI greater than 7, indicating that proteins involved in lipid biosynthesis are generally basic in nature. However, the deduced sequences for genes such as acetyl-CoA carboxylase, acetyl-CoA acetyltransferase, glycerol kinase, ethanolamine kinase and phosphoethanolamine cytidyl transferase were determined to be acidic. These values of isoelectric point (overall charge) will be useful for developing a buffer system for purification of the enzymes by an isoelectric focusing method. Instability Index analyses reveals the presence of certain dipeptides occurring at significantly different frequencies between stable and unstable proteins. Proteins with an instability index less than 40 are predicted to be stable while those with a value greater than 40 are assumed to be unstable. In the present study the high occurrence frequency of unstable proteins may be explained in the context of the recent work of Cao, 85 who observed such a phenomenon in many plants and microorganisms due to the possible inherent feedback mechanism that regulates the optimal level of accumulation of cellular metabolites. The aliphatic index refers to the relative volume of a protein that is occupied by aliphatic side chains (eg, alanine, isoleucine, leucine and valine) and contributes to the increased thermal stability observed for globular proteins. Aliphatic index for the screened proteins ranged from 70.24 to 119.16. The very high aliphatic index for all sequences indicated that their structures are more stable over a wide range of temperature. The GRAVY index indicates the solubility of the protein. The lipid biosynthetic proteins which showed large negative values indicated that these proteins are relatively more hydrophobic when compared to proteins with less negative values.

Distribution of various physico-chemical characteristics of putative proteins encoded by lipid genes in A. thaliana, C. reinhardtii, V. carteri, O. lucimarinus, O. tauri and C. merolae.
The secondary structure of the microalgal proteins involved in lipid metabolism were analyzed by submitting the amino acid sequence to the GOR IV program, which has been experimentally cross validated to have a mean accuracy of 64.4% for the three state prediction. 32 The secondary structure indicates whether a given amino acid lies in a helix, strand or a coil. Secondary structure features of the proteins are represented in Table S3. The results revealed that random coil to be predominant followed by alpha helices and extended strands in the majority of sequences.
GC-content analyses
The variations in the guanine (G) and cytosine (C) content observed between species is one of the central issues in evolutionary bioinformatics. The average GC-content of the lipid biosynthetic genes, as calculated by the Genscan server, was 39.89%, 63.35%, 56.92%, 59.88%, 59.04% and 55.57% for A. thaliana, C. reinhardtii, V. carteri, O. lucimarinus O. tauri and C. merolae respectively. The GC values lie close to the calculated GC-content of the whole genome of the respective organisms under study.86–89 However, a slightly higher GC-content for the gene sequences was observed in contrast to the background GC-content for the entire genome of all the studied species. Among the microalgae, the highest GC-content was observed in C. reinhardtii. The GC-content of C. reinhardtii is also experimentally reported to be higher than that of the multicellular organisms. 90 Comparative analyses of the GC-content of the individual genes revealed minor variations among the microalgal genomes (Fig. 4 and Table S4). The above finding is in congruence with the earlier report stating that eukaryotic genomes vary less in their GC content. 91 Furthermore, GC-content analyses indicated that the genes with high GC-content were also identified to be stable by ProtParam server as compared to genes having low GC-content. This may apparently be due to the fact that GC pair is bound by 3 hydrogen bonds (H-bonds), compared to 2 H-bonds in AT, thus contributing to the greater stability of the gene products. In addition, analyses of individual predicted genes in O. lucimarinus and O. tauri revealed more or less similar GC-content in both the subspecies.

Comparison of the GC-content of lipid biosynthetic genes among five unicellular algae and the vascular plant, A. thaliana.
Motif and domain architecture
A motif is a sequence pattern found conserved in a group of related protein or gene sequences. 34 An exhaustive search of the protein motifs using the MEME program identified 36 core conserved sequences in the lipid biosynthetic genes of microalgae predicted in the present study (Fig. 5). The overall height of each stack indicates the sequence conservation at that position, whereas the height of symbols within each stack reflects the relative frequency of the corresponding amino acid (Fig. 5). The sequence logos showed that majority of the predicted motifs are basically composed of hydrophobic and polar uncharged residues. It is likely that these conserved residues are critical for the catalytic activity of the enzymes and may be involved in substrate binding, direct catalysis, and maintenance of the protein structure. In addition to motif analyses, a detailed comparison of the domain architectures of the gene products at the whole genome level is given in Figure 5. Results indicate that the majority of domains observed in genes involved in lipid biosynthesis are present in all microalgal species under study. Therefore, the critical amino acid residues present in the conserved motif and domain of the lipid genes will certainly act as a framework for better understanding their structure-function relationship.
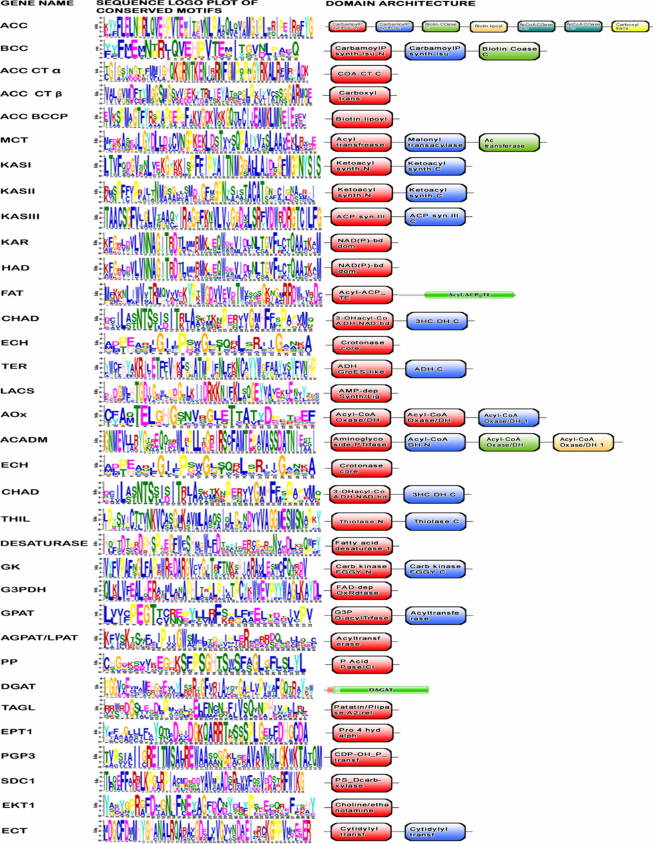
Conserved domain architectures and sequence logo plots of lipid biosynthetic genes using InterProscan and MEME programs, respectively.
Exon-intron structure and phylogenetic analyses
In order to gain insights into the evolution of the lipid biosynthetic genes, we analyzed exon-intron structure patterns of the predicted gene homologs (Table S5). The results revealed that the exon-intron spilt pattern of C. reinhardtii and V. carteri genes were homologous to that of Arabidopsis, although insertion, deletion and intron-size variations were common. Likewise, conservation with respect to exon-intron number and size were observed between O. lucimarinus and O. tauri. The C. merolae genome is remarkable for its paucity of introns 88 and in our study we also could not detect its presence in any of the predicted genes. O. lucimarinus and O. tauri genes contained fewer introns as compared to C. reinhardtii, V. carteri and A. thaliana and our present results confirms the previous report that C. reinhardtii lipid biosynthetic genes contain a higher number of introns. 92 A phylogenetic tree was constructed to evaluate the evolutionary relationship among the predicted genes (Fig. 6). The phylogenetic tree showed that in the majority of predicted genes with similar functions and sharing similar intron-exon structure, conserved motif patterns were clustered together in the tree because of their common ancestry and in accordance with our expectations. In most of the gene families, it was observed that the protein sequence of the two sub-species O. lucimarinus and O. tauri (Prasinophytes) were present as sister clades and that it falls within the green algal cluster comprising of C. reinhardtii, V. Carteri (Chlorophytes) and A. thaliana (Streptophytes). The Chlorophytes and Streptophytes lineages are a part of the green plant lineage (Viridiplantae). 93 Further, the phylogenetic analyses suggest that protein homologs of C. merolae (Rhodophytes) seem to diverge from the root of the green lineage. Overall, we found that components of lipid biosynthetic pathway are remarkably well conserved, particularly among the Viridiplantae lineage.

(A) Phylogenetic tree inferred from the amino acid sequences of lipid genes in A. thaliana, C. reinhardtii, V. carteri, O. lucimarinus, O. tauri and C. merolae. Proteins with identical functional characterization are represented by similar colour coded diamond shapes. Protein accession numbers are represented while organism names to which proteins belong are given in Table 1 Some homologous proteins were omitted to increase clarity of the remaining groups. The tree indicates that proteins with similar functions were clustered together and further, in most of the gene families for instance in desaturase (B), the protein sequence of the two sub-species O. lucimarinus and O. tauri were present as sister clades and falls within the green algal cluster comprising of C. reinhardtii, V. Carteri and A. thaliana, while the protein homologs of C. merolae seem to diverge from the root of the green lineage.
Conclusion
Identification of genes responsible for oil accumulation is a pre-requisite to targeting microalgae for enhanced yields of biofuel precursors using metabolic engineering. A comprehensive computational analyses of the predicted genes of microalgae against Arabidopsis was performed through gene annotation, subcellular localization, physico-chemical characterization, exon-intron pattern, motif/domain organization and phylogenomics studies. The results revealed that although each of the algal species maintains the basic genomic repertoire required for lipid biosynthesis, they possess additional lineage-specific gene groups. Additionally, the extensive sequence and structure conservation of the putative genes indicates functional equivalence between microalgae and Arabidopsis. Phylogenetic analyses demonstrated that genes of lipid biosynthetic pathway from Prasinophytes, Chlorophytes, Streptophytes and Rhodophytes were clustered according to their conserved motif pattern, exon-intron structure and functional equivalence. The in-depth broad investigation of each individual gene and their encoded products across the microalgal genome will certainly facilitate metabolic engineering of microalga for biofuel production.
Author Contributions
Conceived and designed the experiments: NM, PKP. Analysed the data: NM, PKP, BKM. Wrote the first draft of the manuscript: NM, PKP, BKP. Contributed to the writing of the manuscript: NM, BKP, PKP. Agree with manuscript results and conclusions: NM, PKP, BKP, BKM. Jointly developed the structure and arguments for the paper: PKP, BKM. Made critical revisions and approved final version: NM, PKP, BKP, BKM. All authors reviewed and approved of the final manuscript.
Funding
Author(s) disclose no funding sources.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Supplementary Data
Classification of microalgal lipid biosynthetic proteins on the basis of subcellular localization using TargetP, ChloroP and WolfPsort prediction tools.
Footnotes
Acknowledgement
The research fellowship granted to NM by the Government of India's Department of Biotechnology (under a grant-in-aid project) and subsequently a Senior Research Fellowship award by Council of Scientific ' Industrial Research, India is gratefully acknowledged.
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.