
Abstract
P-glycoprotein (P-gp), an efflux membrane transporter, is widely distributed throughout the body and is responsible for limiting cellular uptake and the distribution of xenobiotics and toxic substances. Hundreds of structurally diverse therapeutic agents are substrates to it and it impedes the absorption, permeability, and retention of the drugs, extruding them out of the cells. It is overexpressed in cancer cells and accountable for obstructing cell internalization of chemotherapeutic agents and for developing transporter mediated resistance by cancer cells during anti-tumor treatments. As it jeopardizes the success of drug delivery and cancer targeting, strategies are being developed to overcome P-gp mediated drug transport. This concise review represents a brief discussion on P-gp mediated drug transport and how it hinders the success of various therapies. Its main focus is on various strategies used to tackle this curb in the field of drug delivery and targeting.
Keywords
Introduction
P-glycoprotein (P-gp) is one of the first members of the ATP-binding cassette (ABC) transporter which acts as a physiological barrier by extruding toxins and xenobiotics out of cells.1,2 It is being extensively studied and experimented upon recently and is gaining much importance in numerous researches. P-gp is primarily found in epithelial cells which have the excretory roles including apical surface of epithelial cells lining the colon, small intestine, pancreatic ductules, bile ductules, kidney proximal tubules, and the adrenal gland.3–5 It is also located in the endothelial cells of the blood brain barrier (BBB). 6 The transporter is overexpressed on the surface of many neoplastic cells and restricts cell entry. The role of P-gp is likely to protect these susceptible organs from toxic compounds, preventing them to enter the cytosol and extrude them to the exterior. 7 Thus it also enhances the secretion of metabolites and xenobiotics into bile, urine, and the lumen of gastrointestinal tract. 1 P-gp in human forms a small gene family with two isoforms. The class I isoform (MDR1/ABCB1) is a drug transporter while the class II isoform (MDR2/3/ABCB4) carries out export of phosphatidylcholine into the bile.2,8 A single P-gp molecule can recognize and transport numerous drugs with a wide range of chemical structures, ranging from a molecular weight of 250 g/mol (cimetidine) to 1202 g/mol (cyclosporin). 9
P-gp can extrude a wide range of structurally diverse compounds out of the cells. Hundreds of substrates (usually hydrophobic) interact with this ATP dependent transporter including anticancer agents, immunosuppressants, steroid hormones, calcium channel blockers, beta-adrenoreceptor blockers, cardiac glycosides, among others.2,10,11 Less permeable drugs (weak substrates) may also undergo a substantial extrusion. Thus it contributes greatly in the extrusion of many drugs from the blood into the intestinal lumen. P-gp is also responsible for enhancing the excretion of drugs out of hepatocytes and renal tubules into the adjacent luminal space. Therefore, P-gp can potentially reduce the absorption and oral bioavailability and decrease the retention time of a number of drugs.9,10 Additionally, it has a role in limiting cellular uptake of drugs from blood circulation into the brain while being present in the BBB. 6 P-gp is overexpressed in cancer cells and is responsible for drug efflux in tumors. It prevents cell internalization of chemotherapeutic agents and makes the chemotherapy almost ineffective in many cases (Fig. 1). Hence, this protein is one of the main barriers in cancer treatment by chemotherapy.12,13 A variety of strategies are being developed to overcome the difficulties associated with P-gp in optimum drug delivery. Those include not only inhibition of P-gp, but also various techniques to bypass it.1,4,15,16 These promising approaches for optimizing drug delivery and targeting will be the focus of discussions in this review.
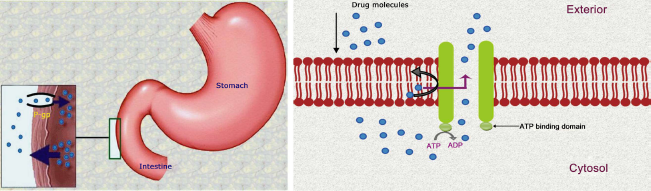
Drug efflux by P-glycoprotein.
Inhibition of p-gp
The inhibition of efflux pump is mainly done in order to improve the delivery of therapeutic agents. In general, P-gp can be inhibited by three mechanisms: (i) blocking drug binding site either competitively, non-competitively (Fig. 2) or allosterically; (ii) interfering with ATP hydrolysis; and (iii) altering integrity of cell membrane lipids.1,10,17–19 The goal is to achieve improved drug bioavailability, uptake of drug in the targeted organ, and more efficacious cancer chemotherapy through the ability to selectively block the action of P-gp. Inhibitors are as structurally diverse as substrates. 19 Many inhibitors (verapamil, cyclosporin A, trans-flupenthixol, etc.) are themselves transported by P-gp. 2
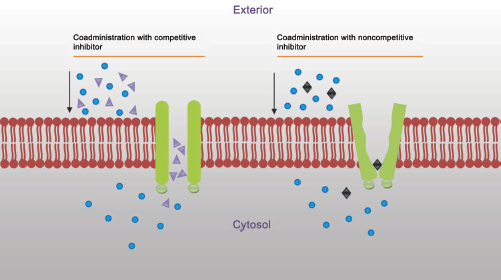
Inhibition of P-glycoprotein to prevent drug efflux.
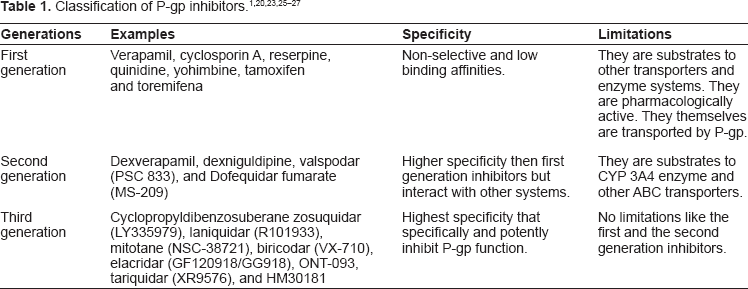
P-gp inhibitors are classified into three generations based on their specificity, affinity, and toxicity. First generation inhibitors are pharmacologically active substances which are clinically used for specific treatments but have the ability to inhibit P-gp. The usage of first generation inhibitors is limited due to their high serum concentrations (at the doses that are required to inhibit P-gp) and potential toxicity.20,21 They are substrate to other transporters and enzyme systems, resulting in unpredictable pharmacokinetic interactions. Second generation inhibitors lack the pharmacological activities and possess a greater P-gp affinity which include non-immunosuppressive analogues of cyclosporin A (PSC833) and D-isomer of verapamil (dexverapamil). However, second generation inhibitors inhibit the CYPA4 enzyme and other ABC transporters. Hence, metabolizing rate decreases and inhibition of two or more ABC transporters lead to complicated pharmacokinetic alterations. Third generation P-gp inhibitors are under clinical development, aiming to inhibit P-gp with higher specificity and lower toxicity (eg, tariquidar). They are developed by using structure activity relationships and many were found to be very specific and effective against P-gp, having minimal toxicity.22–24 Table 1 illustrates the detailed classification of P-gp inhibitors.
Monoclonal antibodies can be effectively used against P-gp in multidrug resistance (MDR) tumor cells. Many anti-P-gp monoclonal antibodies, such as MRK16 and MRK17, have been developed to overcome multidrug resistance. Conjugated monoclonal antibodies, such as bispecific antibody, immunotoxin, and radioisotope conjugates, have also been constructed to enhance the anti-tumor activity of monoclonal antibodies. 28 The conformation-sensitive UIC2 monoclonal antibody can inhibit P-gp mediated substrate transport (calcein-AM, daunorubicin, and 99 mTc-hexakis-2-methoxybutylisonitrile). However, inhibition by UIC2 alone is usually partial because UIC2 binds only 10% to 40% of P-gp present in the cell membrane. This antibody recognizes and inhibits the rest of the P-gp molecules only in the presence of certain inhibitors, including vinblastine, cyclosporine A, and PSC 833 (valspodar). Therefore, simultaneous application of any of these inhibitors and UIC2 can enhance accumulation of certain substrates of P-gp by total inhibition of P-gp pump activity. 29
Enhancement of Bioavailability and Transport
Even though P-gp decreases drug absorption of all of its substrates, it quantitatively does not have an equal significant impact on overall drug absorption for all of them. In the case of fast absorbing drugs having larger doses, efflux by P-gp poses less impact on drug absorption and therefore is not important in terms of bioavailability or pharmacokinetic properties. This is because the transport activity of P-gp becomes saturated by high concentrations of drug in the intestinal lumen. In the case of drugs requiring a very small dose for their pharmacological actions or the drugs that have very slow dissolution and diffusion rates, P-gp mediated drug efflux greatly interferes with their delivery. As it decreases drug absorption, those small amounts of drugs cannot reach the blood circulation in sufficient quantity and, at times, can be life threatening. Moreover, it can make the sustained release dosage forms of the substrates completely ineffective by limiting their absorptions. 9
Usually, an inhibitor of P-gp is coadministered with the drug to enhance drug absorption.30,31 Methods are being developed in preparing clinically useful oral formulations of drugs having poor oral absorption, which are administered only by parenteral routes. When a drug and an inhibitor are coadministered, P-gp mediated transport mechanism remains the same for both the drug and the inhibitor, if the inhibitor is a substrate of it. In this process, drug and inhibitor must be discriminable by P-gp at molecular level as drug molecules should not be extruded. This is why a large difference between the rate of efflux of the substrate and the inhibitor is created. When the inhibitor molecules are effluxed, they rapidly bind to the P-gp binding sites again and thus the inhibition of P-gp by the modulators is cycled repeatedly, preventing efflux of drugs. This process depends on the hydrophobicity of the compounds.32,33
HM30181, a newly developed third generation P-gp inhibitor, showed promising results for increasing oral absorption of some drugs in recent studies. Kwak et al 34 examined its pharmacologic characteristics and it showed the highest potency among several P-gp inhibitors, including cyclosporin A, XR9576, and GF120918. The inhibitory activity of HM30181 was highly selective to P-gp and it did not inhibit other ABC transporters. Its coadministration (10 mg/kg) greatly increased oral bioavailability of paclitaxel from 3.4% to 41.3% in rats. Importantly, oral coadministration of paclitaxel and HM30181 showed a tumor inhibitory strength equal or superior to that of intravenous paclitaxel in the xenograft model in nude mice. 34
P-gp inhibitors may have a great impact on altering pharmacokinetics of a drug. 10 Since P-gp molecules are present in many organs like BBB, kidney proximal tubule, and bile ductule, their inhibition can potentially improve not only the absorption, but also the distribution, metabolism, and elimination of their substrates.10,35 Asperen et al 36 observed a 10-fold increased oral bioavailability of paclitaxel in mice administered along with a P-gp blocker (valspodar). Sugie et al 37 articulated that coadministration of azithromycin and cyclosporine resulted in decreased billiary excretion of azithromycin. BBB is considered as the main barrier to prevent drugs entering the central nervous system (CNS). Presence of P-gp in the BBB makes it additionally difficult, effluxing the drug molecules from the CNS.2,38 P-gp inhibition can prevent P-gp mediated drug efflux and assist the substrate molecules to enter the CNS. Kemper et al 38 reported a 5-fold increase in brain uptake of paclitaxel by GF120918 (elacridar), a third generation P-gp inhibitor. A reduced clearance of paclitaxel was noticed, assuming the inhibition of P-gp in elimination pathway. 38 P-gp inhibition can also increase the half-lives of the substrates as the inhibition may reduce billiary excretion and the clearance of the substrates in kidney proximal tubule, increasing renal reuptake. Orally coadministered doxorubicin and verapamil have shown to increase peak plasma level, prolong elimination of half-life, and increase volume of distribution of doxorubicin after oral administration. 39 Both natural and synthetic inhibitors can be coadministered with drugs in oral dosage forms including polyethylene glycol (PEG) based surfactants, anionic gums, sodium alginate, poloxamers, thiomers, dendrimers, etc. Among these inhibitors, surfactants are considered to be the better choice as they were already approved for routine use in pharmaceutical formulations. These surfactants act by altering integrity of membrane lipids by changing the secondary and tertiary structure. Thus the function of P-gp is hampered due to disturbance in hydrophobic environment.10,40
Antimicrobial therapy
Drug efflux is a common mechanism of resistance in microorganisms, along the same lines as target modification or production of antibiotic inactivating enzymes. Efflux pumps are now recognized as one of the most important factors in accumulation of antimicrobials in all cell types, from prokaryotes to superior eukaryotes. MDR, mediated through efflux pumps, has been described for various organisms, including bacteria, fungi, and protozoa. All transporters, but the ABC family, function as secondary transporters. P-gp is one of the main ABC transporters that is greatly responsible for MDR in microorganisms.41–43 P-gp molecules prevent antimicrobial agents from entering the microorganisms. They reduce intracellular drug concentrations and thereby impede accessibility of drugs to their sites of action, ultimately leading to reduced susceptibility. This transport mechanism may result in treatment failure in many infectious diseases.44,45 MDR is a major problem in bacterial infections because it limits the option of antimicrobial agents. The treatment becomes troublesome in the case of the infections where P-gp substrates are the only choice. It poses several complications including increased costs, prolonged duration of hospital stay, and higher morbidity and mortality rates.46–48 The use of inhibitors may be a good strategy to overcome transporter mediated bacterial multidrug resistance. They can be used in conjunction with antibiotics and can extend the lifetime of the antibiotics, improving therapeutic efficacy. They can also suppress the emergence of resistant variants that may arise during the treatment. These inhibitors increase bacterial susceptibility towards antimicrobial agents.41,49–51
Seral et al 52 examined the influence of inhibitors of P-gp (verapamil, cyclosporine, and GF120918) on the antimicrobial activity of macrolides (erythromycin, clarithromycin, roxithromycin, azithromycin, and telithromycin) in J774 murine macrophages. The result showed that P-gp enhanced the extrusion of azithromycin, erythromycin, telithromycin, and roxithromycin, resulting in suboptimal drug accumulation. Hence, inhibitors can enhance their accumulations inside the cells and increase antimicrobial actions. 52
Leitner et al
49
investigated the potency of tariquidar, a third-generation P-gp inhibitor, for overcoming bacterial resistance towards ciprofloxacin. Activity of tariquidar was evaluated by
Cancer Chemotherapy
P-gp is overexpressed on the surface of cancer cells and prevents drug accumulation inside the tumor, acting as the efflux pump. It extrudes anticancer drugs before they can reach the intended target. Further, it often mediates the development of resistance of the cells to anticancer drugs. Therefore, the administered drugs remain ineffective or cannot bring the desired output. Several approaches have been taken to overcome P-gp mediated drug resistance.53,54 P-gp locates drugs which are localized in the plasma membrane only. Concurrent administration of cytotoxic drugs and inhibiting agents, like verapamil or cyclosporine, can restrain P-gp mediated extrusion and facilitate the drug in reaching the targeted area. Thus both chemotherapeutic agent and inhibiting agent are incorporated into the carrier system to overcome the difficulty associated with P-gp.55,56 Another strategy could be the involvement of anti-P-gp monoclonal antibody in refraining P-gp from extruding drugs. In this process, the antibody is conjugated to the drug loaded carrier system, which can sufficiently inhibit drug efflux.
Vincristine-loaded lipid nanoparticles conjugated to an anti-P-gp monoclonal antibody (MRK-16), showed greater cytotoxicity in resistant human myelogenous leukemia cell lines than non-targeted particles. 14 Goda et al 29 reported that combination therapy with UIC2 monoclonal antibody and cyclosporine A, a first generation P-gp inhibitor administered at the dose ineffective when applied alone, dramatically increased daunorubicin accumulation in xenotransplanted Pgp+ tumors. It established that combined application of UIC2 antibody and a class of modulators, used at low concentrations, can be a specific and effective way of blocking P-gp function in vivo. 29 Danson et al 57 developed SP1049C, a non-ionic block copolymer composed of a hydrophobic core and hydrophilic tail, which contained doxorubicin and was able to circumvent P-gp mediated drug resistance in a mouse model of leukaemia.57,58 Tidefelt et al 59 found that by interacting with P-gp, valspodar can increase the cellular uptake of daunorubicin in leukemic blasts in vivo. In another study, folic acid, attached to PEG derivatized distearoyl-phosphatidylethanolamine in doxorubicin loaded liposomes, was used to target folate receptor overexpressing tumor cells. Folate receptor mediated cell uptake of targeted liposomal doxorubicin into a multidrug resistant subline of M109-HiFR cells (M109R-HiFR) was clearly unaffected by P-gp mediated drug efflux, in sharp contrast to uptake of free doxorubicin. 15
Conclusion
P-gp is one of the main barriers for delivering drugs properly. A variety of approaches are being tested to develop P-gp inhibitors or mechanisms to bypass it. Proper inhibition will allow not only an increase in cellular uptake, transport, and half-lives of drugs, but also to predict their pharmacokinetics accurately and fine tune them for targeting specific region. These advances will result in cost effective therapy by saving the additional amount of drugs that was previously wasted by P-gp transport. Furthermore, it will shorten the treatment time with optimal drug delivery. Thus it can bring a great revolution in the field of drug delivery.
Author Contributions
Conceived and designed the experiments: MLA. Analyzed the data: MLA. Wrote the first draft of the manuscript: MLA. Contributed to the writing of the manuscript: MLA. Agree with manuscript results and conclusions: MLA. Jointly developed the structure and arguments for the paper: MLA. Made critical revisions and approved final version: MLA. Author reviewed and approved of the final manuscript.
Funding
Author(s) disclose no funding sources.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the author has provided signed confirmation of compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.