
Abstract
Gastrointestinal stromal tumor (GIST) is a well recognized and relatively well understood soft tissue tumor. Early events in GIST development are activating mutations in KIT or PDGFRA, which occur in most GISTs and encode for mutated tyrosine receptor kinases that are therapeutic targets for tyrosine kinase inhibitors, including imatinib and sunitinib. A small minority of GISTs possessing neither KIT nor PDGFRA mutations may have germline mutations in SDH, suggesting a potential role of SDH in the pathogenesis. Immunohistochemical detection of KIT, and more recently DOG1, has proven to be reliable and useful in the diagnosis of GISTs. Because current and future therapies depend on pathologists, it is important that they recognize KIT-negative GISTs, GISTs in specific clinical contexts, GISTs with unusual morphology, and GISTs after treatment. This review focuses on recent developments in the understanding of the biology, immunohistochemical diagnosis, the role of molecular analysis, and risk assessment of GISTs.
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor of the gastrointestinal (GI) tract. Prior to their recognition as distinct tumors, GISTs were most commonly classified as smooth muscle tumors or neural tumors. 1 GISTs are now well recognized and well understood by pathologists and clinicians. Our understanding of the biology of these tumors has expanded significantly since the landmark work in 1998 by Hirota and colleagues implicating KIT mutations in the pathogenesis of GISTs, and the subsequent work in 2003 by Heinrich and colleagues uncovering activating mutations in platelet-derived growth factor receptor α (PDGFRA).2,3 The discovery of these tyrosine kinase receptor mutations in GISTs and the fortuitous application of the tyrosine kinase inhibitor (TKI) imatinib mesylate have changed the therapeutic landscape of this previously treatment-refractory tumor. The TKI sunitinib has been approved since 2006 for imatinib-resistant GIST and other TKIs, as well as other therapies, are under active clinical investigation. Treatment of GIST is currently regarded as the paradigm of molecular targeted therapy in solid tumors.
This review provides an overview of recent advancements in the understanding and diagnosis of this tumor; in particular, the role of molecular testing in this era of targeted cancer therapy is discussed.
Epidemiology
Historical data concerning the incidence of GIST is unreliable considering that it was not widely recognized until the late 1990s. Currently, the estimated annual incidence of clinically relevant GIST in the United States is set as high as 6,000 cases. 4 In one study examining the Surveillance, Epidemiology, and End Results (SEER) registry, the incidence is reported to be 0.32 per 100,000 per year, and the prevalence is reported to be 1.62 per 100,00 per year during a 15-year period. 5 However, this value may be an underestimate, as very small GISTs may only be found incidentally at the time of autopsy or when a gastrectomy is performed for other reasons. According to a German and a Japanese study, these micro-GISTs (1 to 10 mm) can be found in up to 35% of patients after the age of 50.6,7 These minute GISTs are immu-noreactive for KIT and often contain an oncogenic mutation in the KIT or PDGFRA gene.6,8 In a recent study, the incidence of GISTs examined prospectively over a two-year period was reported to be higher than that in the SEER registry, at approximately 1.12 per 100,000 per year. However, this study was confined to the Rhone-Alpes region in France, which only represents about 10% of the total French population. 9 The true incidence of GIST may be even higher as diagnostic imaging modalities improve.
GISTs commonly present between the fourth and eighth decades of life, with a median age of approximately 60 years.10,11 Much less commonly, GIST can present in the pediatric population. Overall, there is no clear sex predilection; however, in specific clinical associations, such as those arising in the pediatric population, a female predilection has been noted.12–15 The majority of GISTs are sporadic, but GISTs have been identified in association with neurofibromatosis type 1 (NF1); Carney triad syndrome characterized by a constellation of gastric GIST, extra-adrenal para-ganglioma, and pulmonary chondroma; and Carney-Stratakis syndrome characterized by a constellation of gastric GIST and paraganglioma.13,16–23
Clinical Features
GISTs most commonly arise in the stomach (60%) but can be found throughout the GI tract (Fig. 1). The jejunum and ileum are the second most common site (30%), followed by the duodenum (5%), colorec-tum (4%), and esophagus or appendix (<1%).24–30

Primary GIST anatomic locations and relative frequencies.
Rarely, they can arise in the omentum, mesentery, or retroperitoneum, where they are referred to as extra-gastrointestinal GIST. However, these may represent large tumors where a connection to the bowel wall was not formed.31,32 A single case of a primary “GIST” in the pleura has also been reported recently. 33 The majority of GISTs (70%) present with non-specific clinical symptoms, which vary depending on the size and site of involvement. The symptoms can include bleeding, perforations, and less commonly, obstruction.11,34 Approximately 20% of cases are asymptomatic and are found during endoscopy, surgery, or radiologic studies for other reasons; 10% of cases are detected incidentally at autopsy. 11 Overall, metastases are uncommon and typically only seen in the setting of late-stage disease with the exception of pediatric GISTs, which frequently present with lymph node metastases. Other metastatic sites include the liver, lung, bone, soft tissue, or skin. Metastases are often seen more than 5 years after the initial surgery.35–37
Pediatric gastrointestinal stromal tumors
Pediatric GISTs represent 1%-2% of all GISTs and occur most commonly in the second decade of life, with a predilection in females. These tumors arise almost exclusively in the stomach and frequently involve the lymph nodes. Only 10%-15% of these GISTs harbor KIT or PDGFRA mutations and, thus, the majority of these GISTs fall under the broad rubric of so-called “wild-type” GIST.12–15
“Wild-type” gastrointestinal stromal tumors
Wild-type GISTs are seen primarily in children (approximately 85%-90%)12–15 and in a small percentage of adults (10%-15%) 34 and are characterized by the lack of KIT and PDGFRA mutations. Consequently, standard GIST therapies (ie, imatinib and sunitinib) are less efficacious in this clinical group. Although the pathogenesis is largely unknown, recent studies have uncovered germline mutations involving succinate dehydrogenase (SDH), most commonly in the subunit genes SDHB and SDHC, resulting in a complete loss or reduction in SDH protein.12,13 Loss of SDH protein expression is effectively demonstrated using traditional immunohistochemistry.13,19,38 The clinical features associated with the presence of SDH germline mutations in wild-type GISTs, which accounted for 12% of wild-type GISTs in one study, has not been defined. 13 In adult wild-type-GISTs, BRAF exon 15 V600E mutations have been detected in 7%-13% of GISTs, commonly located in the small bowel.39,40
Gastrointestinal stromal tumors in association with Carney's triad and Carney-Stratakis
GISTs are one of the characteristic tumor types found in Carney's triad and Carney-Stratakis. This clinical subtype has unique clinical features compared with their sporadic counterparts, including occurrence at a younger age, a female gender predilection, multi-focality, slow growth, frequent metastases (similar to pediatric GISTs), lack of response to imatinib treatment, and, infrequently, a fatal outcome. This tumor commonly presents in the gastric antrum and does not harbor KIT or PDGFRA mutations. Interestingly, there is no correlation between conventional risk assessment and tumor behavior; even with metastatic disease, clinical behavior is unpredictable, with many cases having a relatively favorable outcome regardless of clinical stage.22,23,41,42
Neurofibromatosis type 1 associated gastrointestinal stromal tumors
GISTs arising in patients with NF1 typically present with multiple tumors involving the small bowel. The majority of these tumors are mitotically inactive and clinically benign. However, clinically malignant GISTs can arise in these patients. Whether or not these GISTs arise sporadically or represent malignant transformation from a benign tumor is unclear. Interestingly, these tumors generally do not harbor either KIT or PDGFRA mutations. The characteristic loss of neuro-fibromin function (encoded by the NF1 gene) allows hyperactivity of the RAS proto-oncogene, which is downstream of KIT and likely plays a role in the pathogenesis of this tumor.16,21,43
Pathological Description
Macroscopic features
GISTs are well-circumscribed tumors most commonly arising in the muscularis propria of the GI tract. The tumor size varies; for high-risk GIST, the median tumor size is 8.9 cm. 44 These tumors have a fleshy pink or tan-white cut surface with hemorrhagic foci, central cystic degenerative changes, or necrosis.
Microscopic features
GISTs are monotonous tumors that can be divided into three principal subtypes depending on the morphology. The majority of GISTs (approximately 70% of cases) are composed of spindle cells with palely eosinophilic fibrillary cytoplasm, ovoid nuclei, and syncytial cell borders. Paranuclear vacuolization is frequently seen (Fig. 2A and B). Extracellular deposits of dense, collagen (skeinoid fibers) are also seen (Fig. 2C). The cells are arranged in short fascicles or whorls. NF1-associated GISTs seem to invariably show this morphology. 16 About 20% of cases are composed of epithelioid cells with palely eosinophilic to clear cytoplasms and round nuclei. The cells are arranged in nests, sheets, and, less commonly, cords (Fig. 2D). This morphology is commonly seen in pediatric GISTs.12,20 The remaining 10% of GISTs have a mixed spindle and epithelioid cell morphology. Regardless of the cytomorphology, GISTs are variably cellular and can have sclerotic, collagenous, or myxoid stromal changes. Pleomorphism can be seen very occasionally.

GIST subtypes and morphology. (A) GIST composed of spindle cells with paley eosinophilic, fibrillary cytoplasm with focal paranuclear vacuolization, H&E 200X. (B) GIST showing more prominent paranuclear vacuolization, H&E 200X. (C) GIST with spindle cells and extracellular skeinoid fibers, H&E 200X. (D) GIST composed of epithelioid cells with eosinophilic and clear cytoplasm, H&E 200X.
After treatment with TKIs, responsive GISTs may show dramatically decreased cellularity and stromal changes, including marked hyalinization and myxoid features (Fig. 3). 45 Other described post-treatment findings include: loss of KIT expression; a change from spindle to purely epithelioid cell morphology; a pseudopapillary epithelioid growth pattern; and, rarely, rhabdomyosarcomatous differentiation.46,47 These changes can create diagnostic challenges for the unaware pathologist.
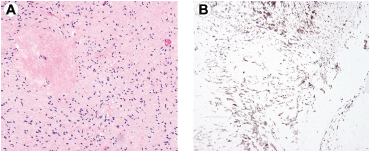
GIST after treatment with tyrosine kinase inhibitors. (A) GIST with decreased cellularity and hyalinized areas, H&E 40X. (B) KIT immunohistochemical staining can be useful in highlighting residual neoplastic cells, 40X.
Oncogenic KIT and PDGFRA mutations
KIT and PDGFRA genes both encode structurally similar tyrosine kinase receptors. These receptors are composed of an extracellular ligand-binding region, a transmembrane sequence, a juxtamembrane domain, and two cytoplasmic kinase domains. In GISTs, mutations in KIT and PDGFRA result in expressed proteins with constitutive oncogenic signaling in the absence of their ligands. The uncontrolled kinase activity results in alterations to cell cycle, protein translation, metabolism, and apoptosis.48,49 KIT and PDGFRA mutations are mutually exclusive.
In GISTs, mutations in KIT and PDGFRA genes generally involve either the cytoplasmic kinase domain or the juxtamembrane (intracellular or extracellular) regions (Fig. 4). The vast majority of KIT mutations are juxtamembrane and found in exon 11 (~70%) and in exon 9 (~10%).3,18 The former consists of variables, deletions, point mutations, and insertions that can involve virtually any portion of the exon; although hot spots do exist with the latter, these almost invariably represent a characteristic duplication of amino acids 502 and 503. Both mutations result in ligand-independent kinase activation. GISTs with KIT exon 11 mutations can occur throughout the GI tract whereas KIT exon 9 mutants occur most frequently in the small bowel.4,26,50,51 Rarely, there are point mutations in exon 13 and 17, both of which encode a portion of the kinase domain and lead to kinase activation. GISTs harboring these mutations are very uncommon, but usually arise in the small intestine. 52 PDGFRA mutations represent a minority of the overall GISTs (less than 10%) and primarily either exon 18 or exon 14. 53 PDGFRA mutant GISTs are generally limited to the stomach, predominantly epithelioid in morphology, and clinically less aggressive.53,54

Diagram of KIT and PDGFRA receptor tyrosine kinases with location and relative frequencies of mutations.
Immunohistochemistry
KIT expression is a specific and sensitive marker for GIST within the standard differential diagnostic setting (Table 1). Over 90% of GISTs are immunoreactive for KIT. 34 Most GISTs show a strong and diffuse cytoplasmic staining for KIT; a minority of GISTs can also exhibit a dot-like or membranous staining pattern (Fig. 5A).2,55–58 The extent and pattern of KIT immunoreactivity has no impact on the likelihood of treatment response. 59 While other neoplasms, such as a subset of melanomas, can also show expression of KIT, an appropriate panel of immunohistochemical stains will avoid diagnostic errors in the vast majority of cases. 58

GIST showing positive membranous immunoreactivity for (A) KIT, 200X; (B) DOG1, 200X; and (C) CD34, 200X.
Immunohistochemistry and molecular findings of GISTs and its differential diagnosis.

Discovered on GIST1 (DOG1) is a promising new marker (Fig. 5B), which has proven in early studies to be a sensitive and specific marker for GISTs.57,60–65 It is immunoreactive in pediatric GISTs and NF1-associated GISTs. Notably, DOG1 stains about one-third of KIT-negative GISTs. 62 Given that up to 5% of GISTs do not express KIT, this marker is especially useful for diagnosis. Rare DOG1 immunoreactivity has been reported in other mesenchymal tumors, including smooth muscle neoplasms and synovial sarcomas.57,60–65
CD34 was an early marker for GIST and is commonly used in the immunohistochemical workup for spindle cell tumors in the GI tract (Fig. 5C). It is less sensitive and specific than either KIT or DOG1, with rates of expression varying from 50%-90% depending on tumor site. 26 In addition to GISTs, CD34 immunoreactivity has been reported in other mesenchymal tumors, including leiomyosarcoma and others that may enter into the differential diagnosis of GISTs. 58
Antibodies to PDGFRA, a tyrosine kinase receptor closely related to KIT, can be employed in cases of KIT-negative GISTs harboring a mutation in PDGFRA. Strong immunoreactivity can be found most commonly in epithelioid GISTs. Its utility is primarily limited by the reports of inconsistent immunohistochemical results when using commercially available antibodies. In addition, immunoreactivity has also been described in other mesenchymal tumors.66,67
Other commonly used markers include caldesmon, smooth muscle actin, desmin, S-100 protein, and keratin, which can be variably immunoreactive in GISTs (Table 1). Notably, caldesmon immunoreactivity is seen in over two-thirds of GISTs;31,58 smooth muscle actin immunoreactivity is seen in less than one-third of GISTs. S-100 protein, cytokeratins, and desmin immunoreactivity are seen significantly less. 58 Fortunately, significant degrees of desmin and S-100 protein reactivity are rare since these raise the diagnostic issue of smooth muscle tumor and nerve sheath neoplasm or melanoma, respectively, and can be confusing, particularly in small biopsies.
Molecular Testing
Clinical history, traditional microscopy, and immunohistochemistry are usually sufficient to establish the diagnosis of GIST. However, in tumors where the diagnosis remains uncertain, real-time polymerase chain reaction (RT-PCR) testing for KIT or PDGFRA gene mutations may be useful. A number of academic centers offer this type of testing, as do private reference laboratories.
In what is more likely to be the future role of molecular testing in GISTs, there has been an increasing trend, primarily at large cancer treatment centers, to employ routine testing for specific mutations to guide initial tumor management. This trend has largely been driven by phase II-III results of clinical trials that show that response to imatinib may be dependent on the specific KIT mutation. Some PDGFRA mutant GISTs show at least partial response to imatinib; however, the most common PDGFRA mutation in GISTs (D842V) confers a complete resistance to the drug.54,68 In KIT mutant GISTs, a mutation in exon 11 was associated with a higher response rate (67%-83%) than a mutation in exon 9 (35%-48%). Conversely, primary resistance to imatinib was also associated with the specific KIT mutation, in particular primary point mutations in exons 13 and 17. KIT exon 11 mutant GISTs were the least likely (0%-5%) to show primary resistance. GISTs with neither KIT nor PDGFRA mutations showed the least treatment response (0%-39%) and the highest primary resistance (23%) to imatinib. Despite these trends, sometimes GISTs with identical mutations in KIT will respond differently to imatinib. Explanations for this are unclear, but may involve variable plasma levels of imatinib between patients and perhaps additional mutations in other genes. Most of the well-validated data for genotype-response correlations is based on imatinib, which is the first-line treatment for GIST; however, it is clear that other members of the ever increasing family of TKIs that target KIT, PDGFRA, and other receptors will have differing efficacies for various mutation types.34,44,69–71
Whereas initially the great majority of GISTs are often highly responsive to treatment with TKIs, acquired resistance is a vexing problem affecting the majority of patients. Mechanisms of resistance most commonly include secondary (acquired) mutations in the KIT kinase domain and rarely KIT/PDGFRA genomic amplifications or activation of alternative oncogenes.3,68,72 Secondary KIT mutations are most commonly single nucleotide substitutions affecting codons in the ATP binding pocket (exons 13 and 14) and the kinase activation loop (exons 17 and 18). These secondary mutations can be detected in up to 83% of patients.69,73 Recent in vitro and in vivo studies demonstrated that sunitinib, a TKI used after imatinib failure, is only effective against secondary mutations located in the ATP binding pocket but not against secondary mutations in the kinase activation loop.74,75 Liegl et al recently demonstrated the substantial inter- and intra-lesional heterogeneity in TKI-resistant mutations in patients treated with imatinib alone or imatinib and sunitinib. In 67% of patients, 2-5 different secondary mutations were detected in separate metastases, and in 34% of patients, two secondary KIT mutations were even seen within a single metastasis. 72 These findings emphasize that testing of secondary KIT resistance mutations in a biopsy specimen will not aid in demonstrating the whole spectrum of resistance mutations. Thus, there are currently no clear recommendations and indications for resistance testing.
Currently, there are no established guidelines for routine KIT or PDGFRA mutational testing. Irrespective of their mutational status, most GISTs are treated with imatinib as first-line therapy; however, this may change in the future. The National Comprehensive Cancer Network (NCCN) and European Organisation for Research and Treatment of Cancer (EORTC) suggest obtaining mutational testing in GISTs that are unresectable or metastatic at presentation, are in young patients, have epithelioid morphology, and have primary resistance to imatinib. Recent prospective clinical trial data shows that patients with KIT exon 9 mutations respond more poorly than those with exon 11 mutations to 400 mg of daily oral imatinib, but that this difference in response is ameliorated when exon 9 mutant patients receive 800 mg imatinib daily. This higher dose generally does not improve response for the standard, imatinib-sensitive exon 11 mutations. 68 This type of data argues strongly for the relevance of genotyping; although, it is currently not known whether genotyping up front to allow immediate higher dosing offers advantages over dose escalation with initial resistance, as has been the current practice. As our understanding of the relationship between genotype and response to various TKIs increases, genotyping will likely become increasingly relevant for therapeutic selection.
Differential Diagnosis
The differential diagnosis of GISTs is influenced by the morphology of the tumor. Both epithelial and other mesenchymal tumors enter into the differential diagnosis.
Spindle cell morphology
The differential diagnosis for spindle cell GISTs consists primarily of other mesenchymal tumors, including leiomyomas, leiomyosarcomas, intra-abdominal desmoid fibromatoses, schwannomas, inflammatory myofibroblastic tumors, and solitary fibrous tumors. 76
Leiomyomas and leiomyosarcomas are the most likely to be confused with GISTs. These smooth muscle tumors are composed of spindle cells with sausage-shaped nuclei, brightly eosinophilic cytoplasm, and distinct cell borders. In contrast, spindle cell GISTs have more ovoid nuclei and have a more syncytial appearance. All 3 tumors can be immunoreactive for smooth muscle actin and caldesmon. Although desmin positivity is rarely seen in GISTs (around 1%-2%), it is much more common in leiomyomas and leiomyosarcomas (greater than 90%-95%).58,76
Intra-abdominal desmoid fibromatoses are composed of myofibroblastic cells set in an eosinophilic collagenous matrix and arranged in long sweeping fascicles. These tumors can be positive for smooth muscle actin and, infrequently, KIT; 77 immunohistochemistry demonstrates strong nuclear staining for β-catenin in 70% of cases.78–80 Ancillary testing for CTNNB1 or APC gene mutations can also be employed. 81
Inflammatory myofibroblastic tumors (IMTs) are composed of atypical myofibroblastic cells arranged in fascicles and admixed with a prominent lymphoplasmacytic infiltrate. In contrast to GISTs, the spindle cells have more vesicular tapering nuclei and more well-defined cell borders. IMTs are immunoreactive to both smooth muscle actin and desmin. In addition, ALK-1 immunoreactivity and ALK gene rearrangement can be seen in a subset of these tumors.82,83
Schwannomas in the GI tract are composed of bland spindle cells with wavy nuclei and fibrillary cytoplasm. The tumor is typically surrounded by a cuff of lymphocytes and diffusely expresses S-100 protein and glial fibrillary acidic protein (GFAP).84,85 Solitary fibrous tumors are also composed of bland spindle cells, but these are arranged in characteristically pattern-less architecture with stag horn vessels. The cells have scant cytoplasm and short, stubby nuclei.
Both GISTs and solitary fibrous tumors can express CD34.56,58,76 However, KIT or DOG1 expression has not been reported in these tumors. 65
Epithelioid morphology
The differential diagnosis for epithelioid GISTs includes both carcinomas, such as neuroendocrine carcinomas and other mesenchymal neoplasms, such as clear cell sarcomas. Neuroendocrine carcinomas are characterized by cytokeratin, synaptophysin, and chromogranin positivity. Clear cell sarcoma, of which a subset occurs as a primary GI tract tumor, expresses S-100 protein and second-line melanoma markers, such as HMB-45 or melan A, and are also characterized by EWSR1-CREB1 or -ATF1 gene fusions.86,87
Predictors of Clinical Behavior
Risk assessment in gastrointestinal stromal tumors
Because the assessment of GIST recurrence risk is currently based on morphology, the pathologist has an important role in the clinical management and optimal care of GIST patients. Numerous studies have shown that mitotic activity and tumor size are highly prognostic of the risk of aggressive primary tumors. 1 These two parameters served as the initial foundation for the consensus approach for risk assessment in GIST. Subsequent studies have added a third parameter, anatomic location, as the next strongest and useful prognosticator. Miettinen and colleagues have shown that small intestinal and rectal GISTs were generally more aggressive than those in the stomach (Table 2).
Risk stratification of primary GISTs based on mitotic index, tumor size, and anatomic site. *

Adapted from Miettinen M, Lasota RJ. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23:70-83, with permission from Elsevier;
denotes small number of cases.
Based on these studies, the 2007 and 2010 NCCN and EORTC guidelines recommend that risk assessment for GISTs be determined by tumor size, anatomic site, and mitotic activity in order to determine which patients will receive adjuvant TKI therapy.34,88,89 Tumor rupture is also associated with an increased risk of recurrence.90–93 However, whether or not tumor rupture is an independent prognostic factor is controversial. Joensuu et al and Rutkowski et al both found that ruptured tumors exhibited more aggressive features; (ie, larger size, increased mitotic figures, and aggressive genotypes).91,92
Conclusion
Although GIST is now a well-recognized entity, the pathologist must be aware of the wide morphologic spectrum of GISTs, including the common morphologic subgroups, the unusual morphologic variants, and the morphologic changes that may be encountered after treatment. Despite these differences in morphology, however, almost all cases can be assessed for risk stratification using the 2007 and 2010 NCCN guidelines along with those from the EORTC, which include mitotic activity, tumor size, and tumor location. In light of the morphologic spectrum, immunohistochemistry for KIT and CD34 among others is exceedingly useful in distinguishing GISTs from their morphologic mimics. Promising new immunohistochemical markers, such as DOG1, will likely be diagnostically valuable, especially in GISTs lacking KIT expression. Currently, there is no standard of care requiring routine molecular testing in situations other than confirmation of the diagnosis in KIT-negative or DOG1-negative cases. However, in the future, molecular testing may become commonplace for guiding initial treatment. It is also likely that the treatment of GIST will evolve toward the simultaneous use of multiple TKIs and perhaps agents that act through other mechanisms in order to avoid the emergence of resistance.
Author Contributions
Analysed the data: WCF, BLA, AJL. Wrote the first draft of the manuscript: WCF. Contributed to the writing of the manuscript: WCF, BLA, AJL. Agree with manuscript results and conclusions: WCF, BLA, AJL. Jointly developed the structure and arguments for the paper: WCF, BLA, AJL. Made critical revisions and approved final version: WCF, BLA, AJL. All authors reviewed and approved of the final manuscript.
Funding
Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals, who reviewed the manuscript for medical accuracy only. We thank Simon J. Slater, PhD, and Jennifer L. Giel, PhD, CMPP, formerly and currently of Evidence Scientific Solutions, part of the UBC-Envision Group, Philadelphia, PA, respectively, for their medical editorial assistance with this manuscript.
Competing Interests
Wai Chin Foo, MD: None. Bernadette Liegl-Atzwanger, MD: None. Alexander J. Lazar, MD, PhD: Novartis, Pfizer, GlaxoSmithKline, Roche, and GE Healthcare.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.