
Abstract
Indoleamine 2,3-dioxygenase 2 (IDO2), a homolog of the better-studied tryptophan-catabolizing enzyme IDO1, is an immunomodulatory molecule with potential effects on various diseases including cancer and autoimmunity. Here, we review what is known about the direct connections between IDO2 and immune function, particularly in relationship to autoimmune inflammatory disorders such as rheumatoid arthritis and lupus. Accumulating evidence indicates that IDO2 acts as a pro-inflammatory mediator of autoimmunity, with a functional phenotype distinct from IDO1. IDO2 is expressed in antigen-presenting cells, including B cells and dendritic cells, but affects inflammatory responses in the autoimmune context specifically by acting in B cells to modulate T cell help in multiple model systems. Given that expression of IDO2 can lead to exacerbation of inflammatory responses, IDO2 should be considered a potential therapeutic target for autoimmune disorders.
Indoleamine 2,3-Dioxygenase 2 (IDO2) and Tryptophan Catabolism
Higher organisms have multiple enzymes that can catabolize tryptophan (Trp, see Table 1 for list of abbreviations), namely, IDO1, IDO2, and tryptophan 2,3-dioxygenase (TDO). Both TDO and IDO enzymes, while unrelated evolutionarily, catabolize the oxidative cleavage of L-Trp to N-formylkynurenine. 1 TDO generally has a narrower expression and substrate specificity, with the majority of its activity in the liver and its substrate constrained to L-Trp alone. In contrast, IDO enzymes are expressed in various tissues and can catabolize a wider range of indole-containing substrates. It is unclear why so many Trp-catabolizing enzymes are necessary or how the convergent evolution of TDO and IDOs occurred, though the wider catalytic range of IDO enzymes and their important link to immunity underscores the differing functions of enzymes that can fundamentally catabolize the same reaction.
Abbreviations used.
There are two closely related IDO enzymes, IDO1 and IDO2, the product of an ancient gene duplication event that occurred prior to the evolution of vertebrates. 2 The two enzymes are closely linked on chromosome 8 in humans (as well as mice) and oriented head to tail. While IDO1 and IDO2 share a high level of sequence identity, they have important kinetic and functional differences. Expression pattern of the two enzymes differs dramatically. IDO1 is expressed in various tissues, whereas IDO2 is expressed in only a subset of these, namely, liver, kidney, and antigen-presenting cells (APCs) of the immune system, including dendritic cells (DCs) and B cells.3,4 Functionally, IDO1 is better able to catabolize Trp to its primary product, N-formylkynurenine, which is subsequently converted to kynurenine. Enzymatic studies demonstrate that IDO2 is significantly less active, particularly human IDO2, though it remains to be seen if the proper substrate and/or enzymatic assay conditions have been correctly identified, as IDO2 activity toward L-Trp and various Trp derivatives is both highly pH and buffer dependent.5–7 Unlike IDO1, which efficiently catabolizes L-Trp, there are several Trp derivatives, particularly 5-methoxytryptophan, that are more efficiently catabolized by IDO2 than Trp itself. 5 Underscoring the murky relationship between IDO2 and Trp catabolism, deletion of IDO2 in mouse models does not alter serum kynurenine levels, even under chronic inflammatory conditions.3,4 Given the nonenzymatic signaling functions defined for IDO1,8–12 it is also plausible that IDO2 has a nonenzymatic function yet to be elucidated, a possibility that must certainly be considered given the generally sluggish catalytic activity of IDO2.
IDO Pathway and Immune Modulation
The IDO pathway was first linked to immunity in a study demonstrating that it is essential in the maintenance of maternal-fetal tolerance. 13 Subsequent work has expanded our understanding of the immunoregulatory role IDO1 plays in modulating regulatory T-cell populations and mediating immune escape in cancer.14–16 Because of this long-standing awareness of the connection between IDO1 and immunity, this connection is significantly better characterized for IDO1 than it is for IDO2. In this review, we focus on the relationship between IDO2 and immune modulation, highlighting key mechanistic differences from those defined for IDO1.
IDO2 has also been implicated in regulatory T-cell differentiation in vitro. 3 Several groups have reported that IDO2 is expressed in DCs and may serve as an immunomodulatory function in this cell type.3,9,17 In human DCs, IDO2 has an expression pattern distinct from IDO1, which is also expressed in DC populations. 17 Similar to IDO1, IDO2 may contribute to immune tolerance by acting in monocyte-derived DCs to control regulatory T-cell (Treg) populations, suggesting an immunosuppressive role, although upregulation of IDO2 was also reported in peripheral blood mononuclear cells (PBMCs) of patients with inflammatory disorders, suggesting that IDO2 may contribute positively to inflammatory processes. 17 Together, these data characterize IDO2 as a complex immunomodulatory enzyme that acts through APCs to affect T-cell populations.
The relationship between IDO2 and immune responses to cancer is less clear. IDO2, which normally has a very restricted range of expression, has been found to be expressed in several cancers, including pancreatic, 18 gastric, colon, and renal tumors. 19 However, not all cancers express IDO2, and some cancers (eg, cervical cancer) actually show down-regulated IDO2 expression compared to normal tissue. 20 Mechanistically, CD8+ T-cell responses against IDO2 have been identified in PBMCs from normal and cancer patient samples, with these cells showing antitumor activity against some, but not all, cancer cell lines. 21 Although IDO2 expression has been identified in several human cancers, preclini-cal carcinogenesis models have yet to show a conclusive direct link between IDO2 and cancer. 3
Connecting IDO and Autoimmunity
Long-standing evidence of reduced tryptophan levels and increased tryptophan catabolites in the serum and urine of patients with autoimmune disease has suggested a role for tryptophan catabolism by IDO.22–26 These initial observations led several groups to examine the contribution of IDO enzymes to autoimmunity, with conflicting results. Several studies have demonstrated an immunosuppressive role for IDO in inducible models of autoimmunity, including models of colitis, collagen-induced arthritis, and experimental autoimmune encephalomyelitis.27–29 Other models, including allergy, contact hypersensitivity (CHS), and inflammatory airway disease, have provided evidence that IDO positively influences inflammatory responses.30–32 Even within a single model, the contribution of IDO can be controversial and dependent on whether small molecule inhibitors or genetic knockouts are used to study its effects.29,33,34
The IDO pathway has been most strongly linked as a positive mediator of autoimmunity through a series of studies conducted using the KRN preclinical model of rheumatoid arthritis (RA). Initial tests of the relationship between IDO and RA used a small molecule inhibitor of the IDO pathway, 1-methyltryptophan (1MT). 30 The widely held belief that IDO1, and by extension IDO2, should suppress effector T-cell functions fostered the expectation that inhibiting IDO activity should exacerbate RA. Surprisingly, the results revealed the opposite to be the case. Administration of 1MT in fact ameliorated disease, with mice demonstrating both delayed onset and reduced severity of disease. 30 This effect of 1MT appears to be mediated not through a reduction in Tregs or altered Th cytokines, but rather from a diminished autoreactive B-cell response, with reduced numbers of antibody-secreting cells and a concomitant decrease in serum autoantibody levels. 30
IDO2 as a Pro-Inflammatory Mediator of Autoimmune Responses
While this work connected IDO to autoimmunity, 1MT likely acts indirectly on the IDO pathway and these experiments could not determine whether IDO1 or IDO2 was responsible for the B-cell-mediated effect on arthritis. To directly examine the relative contributions of IDO1 and IDO2 to the arthritic response, genetic knockouts of each of these genes were bred onto the KRN model of arthritis. Comparison of wild-type (wt), IDO1 knockout (ko), and IDO2 ko KRN.g7 mice (Fig. 1A) revealed that IDO2, but not IDO1, was necessary for robust arthritis development. IDO2 ko mice had a delayed onset and reduced arthritis severity compared to wt or IDO1 ko KRN.g7 mice, which were indistinguishable. The attenuated arthritis was due to a reduction in antibody-secreting cells and corresponding decrease in pathogenic autoantibodies in IDO2 ko arthritic mice relative to their wt or IDO1 ko counterparts. 4 Importantly, the reduced autoimmune response seen in the IDO2 ko mice phenocopies the effect seen with 1MT, suggesting that 1MT can be used as an effective proxy for IDO2 deletion in this system. 30
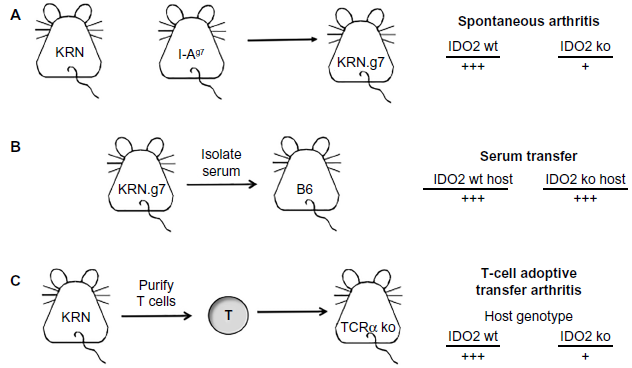
Arthritis can be induced in the KRN model in three different ways:
The arthritic response in KRN mice can be separated into two stages, the B- and T-cell-dependent initiation stage and the downstream effector stage that occurs once autoantibodies trigger activation of neutrophils, macrophages, and mast cells (Fig. 2). 35 To distinguish whether IDO2 was required for the initiation and/or effector phase of the autoimmune response, the B- and T-cell-dependent initiation stage was bypassed by transferring serum-containing pathogenic autoantibodies into wt and IDO2 ko mice (Fig. 1B). IDO2 ko mice developed robust arthritis that was indistinguishable from wt mice, demonstrating that the downstream effector response in IDO2 ko mice is able to mount an arthritic response once pathogenic autoantibodies are supplied. 4 Therefore, IDO2 functions prior to the generation of autoantibodies in the initiation stage of the response.

Model of IDO2 in autoimmune arthritis development. IDO2 acts in B cells to promote the cross talk between B and T cells and the number of antibody-secreting cells. IDO2 is required for robust autoantibody production. Adapted from Ref. 48 with permission from Taylor & Francis Ltd, http://www.tandfonline.com.
Interestingly, IDO2 seems to specifically mediate the autoimmune response but does not affect total B-cell responses. While there is a reduction in autoantibodies in IDO2 ko arthritic mice, total Ig levels remain unchanged between IDO2 wt and ko. Additionally, IDO2 ko mice are able to mount high-affinity isotype-switched B-cell responses after immunization with the model antigen NP (4-hydroxy-3-nitrophenyl acetyl). 4 These genetic studies in mice revealed a specific pathogenic role for IDO2, challenging the general paradigm of IDO function as solely immunosuppressive by suggesting that IDO2 function is distinct from IDO1, operating as a pro-inflammatory mediator in an autoimmune context.
The pro-inflammatory role of IDO2 identified in the KRN model of RA is not confined to this single system. IDO2 has also been shown to be a critical player in CHS responses. 3 In this model, IDO2 ko mice mount a reduced inflammatory response, as defined by ear swelling following a secondary challenge with the hapten oxazolone. While IDO1 ko mice also show a difference in CHS response, the mechanism for the reduction in ear swelling following hap-ten challenge differs between the IDO1 and IDO2 ko strains. Unlike IDO1-deficient mice, IDO2-deficient mice have reduced inflammatory cytokines IL-6, IFN-γ, and TNF-α, hematopoietic cytokines GM-CSF and G-CSF, and reduced MCP-1 relative to wt mice. These data again point to the pro-inflammatory function of IDO2 and identify IDO1 and IDO2 as nonredundant players in inflammatory responses.
In addition to the CHS model, we have recently discovered that IDO2 also contributes to autoantibody production in a mouse model of systemic lupus erythematosus (SLE). Since deletion of IDO2 phenocopies the effect seen with the inhibitor 1MT in the KRN model, we used 1MT as a proxy inhibitor for IDO2 on the MRL/lpr mouse model of SLE, a complex genetic background not amenable to breeding to genetic knockouts. We find that 1MT dramatically inhibits production of anti-dsDNA autoantibodies in this system (Fig. 3). Importantly, this reduction is seen if 1MT is given either before the onset of disease (3 weeks, Fig. 3A), or after (8 weeks, Fig. 3B), demonstrating that 1MT is able to inhibit the secretion of high titers of autoantibodies in MRL/lpr mice in both preventative and therapeutic settings. Of note, this reduction in autoantibodies with 1MT is in contrast to a previous report in the literature. 36 It is not clear why our findings differ, although variances in MRL/lpr mouse colonies may explain this discrepancy; control mice in the study by Ravishankar et al. 36 did not increase their autoantibodies significantly between 8 and 14 weeks of age, a strong difference compared to other MRL/lpr mouse studies, including our own.37–39 Together, these data from SLE, RA, and CHS models provide strong evidence for IDO2 as a mediator of inflammatory autoimmunity in multiple systems.

Treatment of lupus-prone mice with 1MT reduces autoantibody production. MRL/lpr mice were treated with
IDO2 Acts in b-Cells to Affect T-Cell Function
Further studies identified the cellular underpinnings of the reduced autoantibody load, resulting from IDO2 deletion in mouse models of autoimmunity. IDO2 ko KRN.g7 arthritic mice have reduced numbers of differentiated CD4+ T-helper cells and reduced levels of the cytokines IL-4, IL-6, and IL-21, which are involved in the cross talk between B and T cells (Fig. 2). 4 Inflammatory cytokines themselves such as IFNγ and TNFα are not different between IDO2 wt and ko mice. To determine if IDO2 is acting intrinsically or extrinsi-cally to the T cell itself to affect B-cell function, particularly development of antibody-secreting cells, a series of adoptive transfers were performed (Fig. 1C). Here, IDO2 wt or ko KRN T cells were transferred to IDO2 wt or ko T-cell-deficient hosts. These studies revealed that the presence or absence of IDO2 in the host mouse, but not the T cell itself, determines the course of arthritis. 3 IDO2 ko hosts show an attenuated arthritic response, regardless of the genotype of the transferred T cell. This demonstrates that while the functional effect of IDO2 may be mediated through T cells, IDO2 is acting in a cell type extrinsic to the T cell to modulate this response.
To determine which cell type is critical for this IDO2 function, further, more complex reciprocal adoptive transfers were performed. First, to distinguish whether IDO2 is acting in the innate or adaptive immune system, IDO2 wt or ko B and T cells were transferred to Rag-deficient hosts. The host mice have an intact, IDO2 wt innate immune system, but are lacking both B and T cells. We find that only mice that receive wt B cells develop arthritis (irrespective of the genotype of transferred T cells), demonstrating that B cells are necessary for the arthritic response. 40
To determine if IDO2 wt B cells are sufficient for the response, or if IDO2 is needed in other cell types in addition to B cells, a second set of adoptive transfers were performed in which wt or IDO2 ko B cells were added to IDO2 ko T-cell deficient hosts along with IDO2 ko KRN T cells. Thus, all immune components of this mouse, including the endogenous B cells, are IDO2 ko except for the transferred B cells. Here, only the mice with transferred IDO2 wt B cells were able to recapitulate the wt arthritic response. 40 This result was confirmed in the CHS model, where the addition of IDO2 wt B cells was again able to restore the inflammatory response in an IDO2 ko host to levels seen in IDO2 wt mice. 40 Overall, these results offer a new understanding of the contributions of the IDO pathway to chronic inflammation by distinguishing functions of IDO2 and IDO1 as distinct modifiers of immune responses and defining a novel B-cell-specific pro-inflammatory role for IDO2 (Fig. 2).
Given the clear functional role of IDO2 in B cells, further study is needed to define the mechanism by which IDO2 is acting in a B cell to affect differentiated T-cell populations. On a cellular level, adoptive transfer experiments clearly demonstrated that the IDO2-expressing B cell must be cognate and antigen-specific in order to modulate arthritis, suggesting that IDO2 is acting at the direct interface between B and T cells (Fig. 2). 40 Given the observed difference in cytokines mediating B:T-cell cross talk, particularly IL-4 and IL-21, obvious candidates for downstream effectors of the IDO2 pathway are the multitude of co-stimulatory molecules needed for this interaction. Despite examination of a large panel of these markers, including CD80, CD86, ICOSL, IL-21R, OX40L, PDL1, and the MHC Class II molecule itself, the only difference noted is in CD40, which is down-regulated on IDO2 ko B cells. Although an important hint that IDO2 may be affecting co-stimulation, the pathway potentially linking CD40 and IDO2 is not known and remains the subject of active investigation.
Molecular Mediators of IDO2 Function
The molecular mechanism linking IDO2 (as well as IDO1) to immunity remains an area of active research. Because many studies of IDO1 have used the small molecule inhibitor 1MT, which also affects IDO2, it is difficult to clearly interpret the relative contribution of the two enzymes. To specifically define IDO2 function, it is important to determine what regulates IDO2 expression, the downstream mediators of IDO2 activity, and whether, like IDO1, the role of IDO2 in immunity relates directly to catabolic function and amino acid sufficiency/deficiency signals.
IDO2 can be induced in several different ways. In B cells, in vitro assays with purified primary cells demonstrate that IDO2 is upregulated following induction by either a T-independent toll-like receptor (TLR) agonist (lipopolysaccha-ride [LPS] or CpG) or a T-dependent mimic (anti-CD40) when in conjunction with a corresponding cytokine signal, particularly IL-4 or IL-21. 40 The upregulation of IDO2 with LPS + IL-4 has also been reported in macrophages and B cells by others.3,8 While we did not find upregulation of IDO2 by interferon γ (IFNγ) in B cells, IDO2 does appear to be induced by IFNγ in DCs in other systems.19,41 In bone marrow-derived DCs, IDO2 is also upregulated by aryl hydrocarbon receptor (AhR) stimulation. 42 Interestingly, the stabilization of IDO1 and IDO2 transcripts following expression may differ. Unlike IDO1, which contains two functional immunoreceptor tyrosine-based inhibitory (ITIM) motifs, IDO2 contains only one putative, and likely nonfunctional, ITIM motif and is therefore not targeted for suppressor of cytokine signaling 3 (SOCS3)-dependent ubiquitination and subsequent degradation like IDO1.9,17I
IDO1 has been linked to immunity through amino acid sufficiency and deficiency signals, mediated through mammalian target of rapamycin (mTOR) and general control nonderepressible 2 (GCN2), respectively, pathways critical for IDO1-mediated T-cell suppression.8,43 Alternatively, other groups have suggested that the T-cell suppressive effect of IDO expression occurs through the accumulation of Trp metabolites, mediated through binding to AhR and subsequent reprogramming of naïve CD4+ T cells to a regulatory T-cell phenotype.42,44 Whether these pathways are also active in relationship to IDO2 activity is unclear. At first glance, given the substantially lower activity of the IDO2 enzyme in catabolism of L-Trp5–7 and lack of reduction in serum kynure-nine levels in IDO2 ko models,3,4 IDO2 does not seem to be an obvious candidate for action through Trp sufficiency signaling. In support of this, Qian et al. 45 found that while IDO2 expression could suppress both CD4+ and CD8+ T-cell proliferation in vitro, addition of excess Trp was not able to overcome this suppression. There is, however, some evidence that IDO2 can affect kynurenine levels in some contexts.6,7,17
Human IDO2 Polymorphisms
Human IDO2 is significantly less active than the mouse IDO2 enzyme, underscoring the importance of understanding the relationship, or lack thereof, between Trp catabolism by IDO2 and disease.6,45 There is some evidence that human IDO2 may be expressed as different splice isoforms. 6 In human IDO2, multiple out-of-frame stop codons lead to several truncated isoforms. Additionally, the human IDO2 gene has an alternative first exon not found in mouse IDO2. Interestingly, in an in vitro study, deletion of IDO1 caused an increased expression of alternatively spliced IDO2, 3 though the functional consequences of alternative splicing remain unresolved. Human populations have two common IDO2-inactivating polymorphisms. The first, R248W, introduces a mutation at a catalytic residue and generates a ≥90% reduction in catalytic activity in in vitro assays. 6 The second introduces a stop codon at residue 359, leading to a truncated and catalytically inactive protein product. 6 Both polymorphisms are relatively common in human populations, with at least 25% of the population estimated to have a functionally inactive IDO2.6,18 Other non-synonymous variants have been identified from HapMap and other databases. 46 Generally, the contribution of the different isoforms and polymorphisms to immunity, both in the context of cancer and autoimmune disorders, remain undiscovered. However, genetic evidence indicates that inactivating IDO2 polymorphisms may be less prevalent in the African-American population, 6 which may be of relevance to health disparities associated with this ethnic group including disproportionate rates of SLE onset and disease severity. 47 Furthermore, the Y359STOP mutation is slightly associated with a reduced risk of Crohn's disease, 46 supporting the idea of IDO2 as a proinflammatory mediator in an autoimmune environment.
IDO2 Inhibition as a Therapy in Multiple Models of Autoimmune Inflammatory Disorders
The important role demonstrated for IDO2 in preclinical autoimmune models, together with the multiple functional polymorphisms identified in human IDO2, suggests that IDO2 could be an effective therapeutic target to treat inflammatory autoimmunity. However, targeting IDO2 in autoimmune disease has several key challenges. Preclinical studies using the KRN arthritis model suggest that IDO2 functions primarily in the early phase of the immune response by inhibiting the differentiation of the autoreactive B-cells responsible for its initiation. 4 Further supporting the importance of IDO2 in the initiation phase of the response, the inhibitor 1MT has no effect if given after the onset of arthritis; inhibitors must be dosed prior to disease manifestation for the ameliorated response to occur. 30 Therefore, we hypothesized that combining IDO2 inhibition with current RA therapies that either target the effector stage or reinstate the initiation phase could lead to a more effective amelioration of disease. To test this hypothesis, 1MT was combined with two candidate co-therapeutics: (1)methotrexate(MTX), ananti-inflammatory agent that targets the effector stage of disease and (2) anti-CD20 Ig (rituximab), a monoclonal antibody that depletes the B-cell repertoire, effectively rebooting the immune system.48,49 Treatment with MTX was able to partially inhibit arthritis in K/BxN mice when used as a monotherapy; however, combining MTX with 1MT was significantly more effective than either treatment alone at delaying the onset and alleviating the severity of joint inflammation. 48 Likewise, administration of 1MT following anti-CD20 treatment led to reduced numbers of autoantibody-secreting cells and prevented the flare of arthritis observed in mice treated with anti-CD20 alone following the repopulation of B cells. 49 Together, these data suggest that inhibition of IDO2 is a potential therapeutic candidate for use in combination with MTX or B-cell-depletion therapy to increase their efficacy in the treatment of RA.
A second challenge to the development of IDO2 therapy lies in the problem of specifically targeting this intracellular antigen. To inhibit IDO function, most studies in the literature, including those from our laboratory, have historically used the compound 1MT. However, 1MT is not a direct inhibitor of either IDO1 or IDO2, but rather an indirect inhibitor of the overall IDO pathway.4,8 To date, there are no small molecule inhibitors that specifically target IDO2 but not IDO1 and do not also have additional physiological effects. 50 Targeting strategies using monoclonal antibodies offer a potential alternative option, due to their exquisite specificity. Although generally used to target easily accessible surface-expressed or secreted factors, recent evidence suggests that monoclonal antibodies can also be effective at targeting intracellular antigen (Ag), 51 a strategy we are actively pursuing to specifically target IDO2.
Conclusions
Current evidence reveals IDO2 to be an immunomodulatory enzyme that acts in B cells to modulate autoimmune disease. Although its enzymatic function is poorly characterized, it is clear that the mechanism of immune modulation by IDO2 is distinct from its better-studied homolog, IDO1. IDO2 acts as a pro-inflammatory mediator in multiple models of autoimmune inflammatory disorders, including RA, CHS, and SLE. Because IDO2 is acting to promote inflammation, it is an obvious candidate for therapeutic targeting for treatment of these diseases, particularly in a co-therapeutic setting.
Author Contributions
Conceived and designed the experiments: LMFM, LMN. Analyzed the data: LMFM, LMN. Wrote the first draft of the manuscript: LMFM, LMN. Contributed to the writing of the manuscript: LMFM, LMN. Agree with manuscript results and conclusions: LMFM, LMN. Jointly developed the structure and arguments for the paper: LMFM, LMN. Made critical revisions and approved final version: LMFM, LMN. Both authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgments
We would like to thank Samantha Grabler and Elizabeth Pigott for their contributions to the scientific work discussed in this review and Anita Varkey for comments on the manuscript.