
Abstract
One of the dreaded complications of long term analgesic intake is nephrotoxicity characterized by chronic interstitial nephritis and papillary necrosis. Much of the literature of its epidemiology dates back to 1960s and its impact on present day society is not well documented. Non steroidal anti inflammatory agents reduce pain by blocking prostaglandin generation. Prostaglandins have renal vaso dilatory effects in states of volume depletion to counteract the vasoconstrictive pressor hormones. Earlier analgesic tablets contained a mixture of aspirin, phenacetin and caffeine. Phenacetin and its metabolites have nephrotoxic potential and incidence of analgesic nephropathy was brought down in countries where it was banned. The concentration of phenacetin and its metabolite acetaminophen is increased at the tip of renal papilla due to counter current concentrating mechanism of the loop of henle. These are potent oxidants leading to cell injury due to lipid peroxidation, though their effects are normally counterbalanced by anti oxidant glutathione. Glutathione deficiency at the medulla can be precipitated by co ingestion of aspirin. The exact dose of analgesics which needs to be ingested is unclear but a daily ingestion of 5–8 tablets over 5 years results in clinical nephrotoxicity. The histopathology is one of chronic interstitial nephritis with renal fibrosis. Clinically the patient presents with polyuria, asthenia and anemia. The diagnosis is suspected in a patient with progressive chronic kidney disease without proteinuria. CT imaging of the kidneys show irregular scarred kidneys with papillary calcification and necrosis. Recently, COX-2 inhibitors are promoted as renal safe drugs, but may not be so given the multiple case reports of renal toxicity in post marketing surveys. The treatment of analgesic nephropathy includes discontinuation of offending drug, protein restricted diet, control of blood pressure and statins. In conclusion analgesic nephropathy is a preventable cause of chronic kidney disease and both the patients and treating physicians should be mindful of the potential nephrotoxcity of nonsteroidal anti inflammatory agents when administered for prolonged periods without monitoring renal function.
Introduction
Relief from everyday bodyaches and pains is a universally sought after remedy of mankind as evidenced by the burgeoning market share of analgesics and nonsteroidal antiinflammatory drugs (NSAIDs) as over-the-counter (OTC) drugs. Commonly recognized adverse effects of chronic analgesic intake focus on gastrointestinal and cardiovascular risks. However, there is also a well-established risk between chronic analgesic intake and development of kidney disease. The National Kidney Foundation defines analgesic nephropathy (AN) as “a disease resulting from the habitual consumption over several years of a mixture containing at least two antipyretic analgesics and usually codeine or caffeine. It is characterized by renal papillary necrosis and chronic interstitial nephritis that leads to insidious onset of progressive kidney failure”. 1 Therefore, the development of AN is an important, but completely preventable, cause of chronic kidney disease (CKD). This review serves as a timely reminder about the silent renal threat posed by long-term intake of analgesics.
Epidemiology
The term AN was first introduced in the literature nearly half a century ago. In the 1950s, studies from Sweden and Switzerland demonstrated renal papillary necrosis developing from the consumption of large amounts of phenacetin-containing analgesic mixtures. 2 During the 1960s and 1970s, phenacetin was singled out as the nephrotoxic agent in the analgesic mixtures, leading to its ban in several countries. Thereafter, the incidence has declined markedly.
In the 1980s, studies from Australia showed that 10%–20% of patients admitted in dialysis units were suffering from AN. After the ban on the sale of mixture of analgesics containing phenacetin, the incidence has declined. Extensive literature has been published linking analgesic intake and the development of CKD. 3 Inspite of these studies, the exact dose of analgesics, duration of consumption, and the type of analgesics associated with the development of AN have not been defined. A study from Belgium by De Broe and Elseviers 4 defined analgesic abuse as the consumption of 3000 tablets or more of an NSAID over a five-year period. A Swedish study enrolled 926 patients with newly diagnosed CKD and compared them with 998 control subjects. The odds ratio for the development of CKD was 2.2 (95% confidence interval, 1.4–3.5) in those who were regularly taking aspirin with acetaminophen compared to aspirin use alone. 5 In contrast, a case–control study published in 2007 (907 cases and 3622 controls) studied patients with end-stage renal disease, who were younger than 50 years, from 153 dialysis centers in Germany and 17 centers in Australia and compared them with age- and sex-matched neighborhood controls. They could not find an increased risk of end stage renal disease (ESRD) (odds ratio, 0.8) in patients with high consumption of analgesic mixtures compared to those with no or very low analgesic intake. 6 The differing results from these two studies could be due to the fact that the Swedish study was conducted at an earlier era on older patients (age > 50 years) compared to the German study. Recently, COX-2 inhibitors are promoted as being renal safe with weak evidence base. But individual case reports do highlight the potential for renal toxicity of these drugs. 7 After a flurry of activity in this field in the 1960s, there has been a marked paucity of published literature. In fact, a recent autopsy study carried out in Basle has shown that the classic AN has virtually disappeared from the clinical scene in the 21st century. 8 Nevertheless, there are subtle forms of AN that the clinicians will do well recognize.
Mesoamerican nephropathy (MeN) is a recently discovered entity of CKD that is identified among male sugarcane workers working in hot and humid climates. 9 The hypothesis for MeN is that repeated heat stress and dehydration predisposes these workers to CKD. 10 A common symptom of MeN is back pain or loin pain referred to locally as “chistata” for which the workers consume NSAIDs. 10 A study by Herrera et al on sugarcane workers of El Salvador showed the consumption of NSAID in 41% of their patients. 11 NSAIDs alter the renal hemodynamics causing ischemia, which along with the heat stress and repeated dehydration causes progressive renal injury and fibrosis, leading to the development of CKD. This is further compounded by high fructose-containing drinks, which the workers consume to keep themselves hydrated.10,12
Nephrotoxicity of Individual Drugs
In an experimental study in rats, the incidence of papillary necrosis was studied after high doses of a single analgesic such as aspirin, acetaminophen, or phenacetin. It was seen that the incidence of papillary necrosis was least with phenacetin. It was further shown that the metabolite of phenacetin is acetaminophen, which was responsible for the development of AN. 4 This could explain why the incidence of AN did not change despite the ban on the sale of phenacetin in some countries. The nephrotoxicity of acetaminophen is further enhanced by aspirin as elaborated in the “Pathophysiology” section. The nephrotoxicity of phenacetin is dose dependent. An intake of 6–8 tablets per day for a period of 6–8 years leads to the development of AN. 13
The other combinations of analgesics associated with the development of AN were aspirin and a pyrazolone, acetaminophen and a pyrazolone or two pyrazolones, which were combined with caffeine, codeine, or both. The pyrazolones included antipyrine, salipyrine, aminopyrine, and dipyrone. 4
Sulindac was previously considered to be renoprotective since its hepatic metabolite sulindac sulfoxide had shown the least effect on the renal cyclooxygenase system, whereas the sulfide metabolite is active as a vasoconstrictor. The prodrug of sulindac can, however, cause vasoconstriction and lead to vasoconstrictive renal failure usually after days or weeks. Hence, sulindac is also implicated in AN. 14
COX-2 Selective Inhibitors
COX-2 exists constitutively in renal tissues, especially in the cells of the thick ascending loop of Henle, macula densa, and in renal medullary interstitial cells. Prostaglandins (PGs) generated by COX-2 are involved in tubuloglomerular feedback mechanism, leading to afferent arteriolar vasoconstriction. Constitutive COX-2 expression was demonstrable in renal tissues obtained from postmortem studies of cancer patients and was found in peritubular capillary endothelial cells. This underscores the importance of COX-2 inhibition, leading to renal vasoconstriction in volume-depleted states. 15 Several case reports of celecoxib and rofecoxib (selective COX-2 inhibitors) induced acute kidney injury (AKI) have been published in the last decade. In each one of them, the comorbid condition of the patients was a critical factor that increased the nephrotoxicity, viz., sepsis, volume depletion, CKD, and chronic liver disease. 15 Hence, the same precautions need to be taken for COX-2 inhibitors as is necessary for nonselective NSAIDs.14,16
Pathophysiology
AN is characterized by renal papillary necrosis and chronic interstitial nephritis. The renal injury due to analgesics is mainly in the renal medulla. Burrell et al studied the histopathological changes of AN in Fischer 344 rat models. 17 The earliest changes comprise of thickening of the vasa recta capillaries along with patchy areas of tubular necrosis, indicating that the vascular endothelial cells are involved first. 18 This is followed by papillary necrosis, secondary cortical necrosis, interstitial inflammation, and fibrosis. Phenacetin gets metabolized into acetaminophen and other intermediates, which cause lipid peroxidation that injures cells. 15 As a result of countercurrent mechanisms, there is maximum accumulation of the metabolites at the renal medulla, causing the initial injury to occur at the papillary tip. 20
Aspirin potentiates the nephrotoxicity of both acetaminophen and phenacetin. Acetaminophen undergoes oxidative metabolism by prostaglandin H synthase to form reactive intermediates. These intermediates get detoxified through conjugation by glutathione. If acetaminophen is consumed alone, the glutathione in the papillae is sufficient to detoxify the reactive intermediates. If acetaminophen is ingested along with aspirin, the aspirin gets converted to salicylate, which depletes glutathione in both the cortex and papillae of the kidney, causing the reactive intermediates to damage the renal papillae by lipid peroxidation of tissues.19,20
Nephrotoxicity of NSAIDs
NSAIDs cause nephrotoxicity that can present as various renal syndromes such as acute kidney injury, nephritic syndrome, interstitial nephritis, and chronic renal failure. 21 The common link between these various syndromes is the disruption of PG synthesis. A review of the metabolism of PGs is given below.
PGs belong to the class of eicosanoids derived from the oxygenation of arachidonic acid by cyclooxygenases (COX-1 and COX-2). The first PG to be formed is prostaglandin G2 (PGG2), then PGH2, and followed by PGI2, PGE2, and PGF2 (Fig. 1).
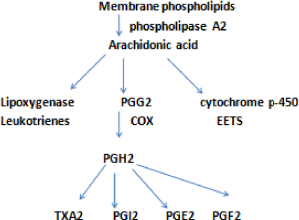
Arachidonic acid pathway for PG generation. 21
The PGI2 has predominant vascular actions in the form of renal vasodilatation. PGE2 has renal tubular action in the form of inhibition of salt and water reabsorption, especially in the thick ascending limbs of loop of Henle and collecting ducts. These biological actions of the autocoids on the kidney are prominent in the setting of a decreased effective arterial blood volume during which there is an increase in the circulating levels of angiotensin II (AII), arginine vasopressin (AVP), and catecholamines. PGs once released act to counterbalance the effect of the abovementioned hormones by causing renal vasodilatation and inhibition of salt and water reabsorption. So the inhibition of PG synthesis by NSAIDs (COX inhibitors) leads to the unopposed action of AII, AVP, and catecholamines, resulting in enhanced renal vasoconstriction and salt and water reabsorption. As the renal medulla is dependent on the production of PGs for its blood flow, the inhibition of PG synthesis by NSAIDs leads to medullary ischemia and papillary necrosis. 21 This is shown in Figure 2.
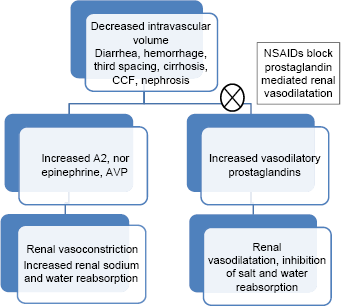
Mechanism of renal toxicity of NSAIDs. 4
Clinical Features
Diagnosis of AN is difficult as the disease has nonspecific symptoms and signs and progresses slowly. Patients may also deny history of chronic analgesic intake. It is important to ask for history of chronically painful conditions like arthritis, headaches, and back pain as these groups are vulnerable to chronic NSAID intake, providing a clue for diagnosis. Patients present with features characteristic of chronic interstitial nephritis, ie, impaired urinary concentrating ability, impaired urinary acidification, and salt-losing state. Proteinuria is usually mild to moderate, which is consistent with chronic interstitial nephritis (CIN). Sterile pyuria is common. The sensitivity and specificity of nonspecific signs such as hypertension, anemia, sterile pyuria, bacteriuria, and proteinuria were found to be insufficient for the diagnosis of AN in patients with end-stage renal disease. 4 Hematuria if persistent should raise the possibility of an underlying uroepithelial tumor.
Diagnosis
Early diagnosis of AN is important as it is a potentially preventable cause of CKD and stopping of analgesics can retard the progression of CKD. Hence, regular screening of serum creatinine and urine dipstick analysis is advised for patients on chronic NSAID intake. As demonstrated by Elseviers et al, 22 noncontrast abdominal computed tomography (CT) scan is useful in diagnosis. In advanced stages, partial or total papillary necrosis with calcification is demonstrated in 25%–40% of patients, which can be demonstrated by a noncontrast CT. Elseviers et al 22 showed that CT findings of a small kidney (sensitivity 95%, specificity 10%) with an irregular or bumpy outline (sensitivity 50%, specificity 90%) and medullary calcification (sensitivity 87%, specificity 97%) with renal impairment and background history of analgesic intake to be suggestive of AN. From these findings, the diagnostic sensitivity and specificity were 92% and 100%, respectively, for detecting AN. These constellation of findings–-small, indented, and calcified kidneys–-were named as “SICK” classification. However, data from the US Analgesic Nephropathy study showed that these findings (SICK) were infrequent in their population and hence are not sensitive to detect AN. 23 A renal biopsy is not useful in a patient who has AN as it shows only nonspecific features such as chronic interstitial nephritis and chronic tubulointerstitial fibrosis.
Prevention and Management
It is one of the few preventable causes of CKD, and early stoppage of NSAIDs can prevent progression of AN. In those with a mild reduction in glomerular filtration rate (GFR) (GFR between 30 and 50 mL/minute), stoppage of NSAIDs prevented further decline in GFR. Studies by Sander et al 24 and Pommer et al 25 showed that habitual consumption of NSAIDs influences the progression of CKD. They also showed that the odds ratio for the development of CKD was higher in those who had interstitial nephritis and nephrosclerosis on renal biopsy. Management of CKD should proceed along the established lines such as protein restriction [0.6 g/kg/day], blood pressure control [below 140/90 mm Hg], use of renin–angiotensin pathway inhibitors, and statins. There are no published studies targeting specific pathways in AN such as antioxidant- and PG-generating drugs. This is a potential yet unmet need in this field.
Conclusion
AN develops due to prolonged and excessive consumption of combination of analgesics and is characterized by renal papillary necrosis, calcification and chronic interstitial nephritis. Though the prevalence of AN is decreasing, it still remains an important preventable cause of ESRD, especially in the developing countries where combination pills are still being manufactured and sold. Screening of patients at a high risk for developing AN is vital for halting the progression to ESRD. Laws to restrict the sale of OTC painkillers will also help in reducing the prevalence of AN.
Author Contributions
Conceived and designed the experiments: KS. Analyzed the data: KS, AR. Wrote the first draft of the manuscript: AR, DS. Contributed to the writing of the manuscript: DS. Agree with manuscript results and conclusions: KS, AR, DS. Jointly developed the structure and arguments for the paper: KS, AR, DS. Made critical revisions and approved final version: KS. All authors reviewed and approved of the final manuscript.