
Abstract
Multiple sclerosis is the most common progressive and disabling neurological condition in young adults. Neuro-inflammation is an early and persistent change and forms the basis of most pharmacotherapy for this disease. Immunomodulatory drugs are mainly biologies (β-interferons, a four amino acid peptide, and a monoclonal antibody to a cell adhesion molecule on the blood-CNS barrier) that either attenuate the inflammatory response or block the movement of immune cells into the CNS. They reduce the rate of relapse, but have little or no effect on the progression of disability. The market landscape for MS drugs is in the midst of major change because the patent life of many of these medicines will soon expire, which will lead to the emergence of biosimilars. In addition, new small molecule immunomodulatory and palliative drugs have entered the market, with more in the pipeline; a number of monoclonal antibodies and other immunomodulatory drugs are also in clinical development.
Keywords
Introduction
Multiple sclerosis (MS) is a disabling disease of the brain and spinal cord that is characterized by multi-focal demyelination and neuronal loss, along with, activation of glial cells and the movement of immune cells into the CNS. It constitutes the most common, non-traumatic, disabling neurological disease of young adults, affecting approximately 2.1 million people worldwide.1–3 The disease is associated with a substantial economic burden, which is attributable to the direct medical costs associated with health along with disability-related resource utilization and indirect costs relating to reduced productivity; there are also indirect costs associated with reduced health-related quality of life. 4 The clinical features of MS are heterogeneous, with its severity ranging from benign to malignant. Symptoms include changes in motor function (particularly walking, manual dexterity, and bladder control), sense perception and mental function (particularly affect and cognition).1–3 Fatigue is a further symptom, which is commonly reported and often initiated by heat. 5
MS presents in different forms that follow different patterns of evolution and rates of disability progression. These forms include: relapsing-remitting (rrMS), secondary progressive (spMS), progressive relapsing (prMS), primary progressive (ppMS) and progressive relapsing (prMS). 6 RrMS is characterized by acute attacks followed by complete or partial recovery with little, or no, disease progression between relapses. PpMS has no periods of remission and the symptoms progress gradually from onset.3,7 Clinically isolated syndrome (CIS) is a precursor to rrMS that usually manifests as a transient impairment in motor or sensory function, along with white matter abnormalities visualized by magnetic resonance imaging, but with insufficient evidence for a definitive diagnosis of MS. 8 In most cases (80% after 20 years), CIS progresses to a diagnosis of rrMS, 9 and the majority of those with rrMS convert to spMS within two or three decades.10,11 PrMS is characterised by a steady neurologic decline and clear superimposed exacerbations. 6 RrMS is the most common form of the disease, followed by spMS, then ppMS (which affects 10%-20% of patients); prMS affects the smallest minority of people with MS (<5%). 6 On the basis of a large longitudinal study, it has been suggested that the various subtypes of MS reflect different clinical phenotypes of a unitary disease. 12
The first clear description of the disease was made by the British physician and pathologist Robert Carswell in 1838. A more complete description followed in 1868 in a series of lectures entitled ‘sclerose en plaques’ from the French neurologist Jean-Martin Charcot. He described a young woman with an unusual tremor, slurred speech and abnormal eye movements. Examination of her brain post-mortem revealed multiple scars or ‘plaques’ that are now recognised as the principal histological hallmark of MS. Both clinical and pathological changes were attributed to the associated loss of myelin.13,14 However, there was little research into MS for about a century after Charcot's description of the disease. This has now changed, with MS firmly established as a major focus of biomedical research. Even so, the cause of MS is not yet known, drugs to significantly slow the progression of disability have not yet been discovered and a cure of this debilitating disease remains elusive.
The development of effective medicines for MS is critically dependent upon understanding the biological basis of this disease. What is clear is that it is a complex multifactorial disease that is influenced by genetics, gender, Epstein-Barr virus (EBV) seropositivity, cigarette smoking and vitamin D3. Despite an incomplete understanding of how these factors interact and how the disease begins and progresses, good progress has been made in the pharmacotherapy of MS. In this article I describe the biology of MS, review current pharma-cotherapeuetic options for its treatment and consider how these options are likely to change in the future.
The Biology of MS
MS is probably an acquired autoimmune disease that is triggered by environmental factors in genetically susceptible individuals. 3 The environment plays a major role since MS concordance rates in genetically identical twins are only about 30% 15 and discordant monozygotic twins do not have significant sequence differences in their genome, epigenome or transcriptome. 16 Even so, the twins cease to be genetically identical as the immune system develops 17 Genome-wide association studies (GWAS) indicate that the strongest genetic risk factor for MS is the HLA-DRB1*1501 haplotype which is located in the class II major histocompatibility (MHC) genomic region on chromosome 6. 18 It encodes the most prevalent beta subunit of human leukocyte antigen (HLA)-DR. Numerous other MS susceptibility loci have been identified from both MHC and non-MHC regions of the genome, but immunologically relevant genes are over-represented among those mapping close to the identified loci. Although the mechanisms by which the associated variants exert their effects on the phenotype of MS are poorly understood, the GWAS data particularly implicates T-helper-cell differentiation and several immunological pathways, including those involving interleukin (IL)-4, IL-6, IL-17 and glucocorticoid receptors.19–21
Both the incidence and prevalence of MS appear to be increasing, with a higher penetrance among women. Thus, the current global female/male ratio of 2 (range: 1.5-2.5) looks set to increase in the years ahead.22,23 A study of gender-dependent inheritance of a genetic locus has identified the MHC (particularly HLA-DRB1*1501) as the site of interactions and modifications mediating the female-to-male gender ratio in MS and its progressive increase. 24
The geographic distribution of MS displays a remarkable latitude effect, whereby there is a north to south gradient of increasing disease incidence; the gradient is the other way around in the southern hemisphere. This finding has recently been substantiated by a comprehensive systematic review of MS prevalence and latitude globally. 25 It strongly supports a role for environmental factors which vary with latitude, the most likely candidate being ultraviolet radiation. This, in turn, determines the concentration of vitamin D3 in the body since it is synthesized from 7-dehydrocholesterol in the skin following ultraviolet light exposure. 1α,25-dihydroxyvitamin D3, the active form of vitamin D3, plays an important role in bone metabolism and can regulate cell proliferation and differentiation as well as apoptosis and immune regulation in immune cells such as T helper cells and dendritic cells. 26 Its mechanism of action is not yet clear, although it has recently been proposed that it acts, at least in part, through an effect on N-glycosylation reactions. 27 In addition, a fascinating interplay between genetics and the environment has recently emerged with the discovery that rare variants in the CYP27B1 gene, which encodes the vitamin D3 metabolizing enzyme 5-hydroxyvitamin D3 1-alpha-hydroxylase, are associated with MS. 28 In addition, a randomised, double blind, placebo controlled trial with vitamin D3 as an add-on to IFNβ-1b in people with rrMS indicated that the vitamin reduced MRI disease activity. 29
There are also pronounced differences in risk among people of common ancestry who migrate to areas of low or high MS prevalence. People who are younger than 16 years at the time of migration tend to adopt the MS risk of the country to which they migrate, whereas those who are older than 16 years have a risk of MS that is similar to their country of origin. 30 Similarly, the emergence of MS among the black population of the French West Indies, where MS has been rare, appears to be attributable to environmental factors that operate in a critical way before the age of 15 years. 31 Thus, childhood is a risk period for the development of MS in adulthood. The most parsimonious explanation is childhood infection such as EBV, which is responsible for infectious mononucleosis, which is characterized by fever, sore throat and fatigue. 32 Further evidence for infectious agents playing a role in MS derives from studies in experimental animals. Thus, transgenic mice expressing genes encoding a rearranged T cell receptor specific for myelin basic protein (MBP) developed experimental allergic encephalomyelitis (EAE) following immunization with MBP and adjuvant plus pertussis toxin, as well as with administration of pertussis toxin alone. The animals were divided into two colonies: one was housed in a non-sterile and the other in a sterile, specific-pathogen-free environment. Nearly all mice in the non-sterile environment were susceptible to a paralytic disease reminiscent of MS, while those in the sterile environment were nearly totally resistant to disease. 33
The data described above, together with MRI studies on the distribution of white matter lesions over time and space and neuropathologic data, indicate that MS lesions are associated with focal perivascular white matter infiltration by T and B lymphocytes in the brain and spinal cord. This strongly supports the hypothesis that MS is a chronic inflammatory disease of the CNS system.34–36
The cascade of inflammatory change associated with MS is probably initiated by autoreactive T cells, particularly CD4+ and CD8+ T helper cells (Th). CD4+ cells recognize antigens that are presented by MHC molecules on specialized antigen-presenting cells. 37 In humans, such molecules largely derive from the classic MHC class I loci HLA-A, -B, and -C and the MHC class II loci HLA-DR, -DQ, and -DP. 30 The key function of these molecules is to bind peptide fragments (epitopes) from foreign antigens, or apparently foreign auto-antigens, and present them to specific T cell receptors. Once primed to attack self in this way, the CD4+ cells switch from physiological surveillance for foreign (usually infectious) agents to pathological destruction of cells of self, myelinated CNS neurons in the case of MS. The most prominent candidates for auto-antigens are proteins present in myelin, such as MBP, myelin oligodendroglial gly-coprotein and proteolipid protein. Other candidates include stress proteins such as B crystallin, which is found in the myelin sheath after activation via the inflammatory response.38,39 However, it is not clear if this is a primary reaction or an epiphenomenon arising during the course of the disease. And if it is a primary event, then how do these proteins cross the blood-CNS barrier to enter the bloodstream?40,41
It is now known that some key components of myelin are present in sites external to the myelin sheath, in places outside the CNS. Thus, MBP is expressed in the thymus during the development of the immune system and into adulthood.42,43 This may be the antigen responsible for the induction and pathogenesis of MS. An alternative hypothesis is that the triggering antigen is foreign in origin. Recent epidemiological and immunological studies indicate an association between EPV infection and MS. EBV is a ubiquitous human herpes virus with the unique ability to infect, activate, and latently persist in B lymphocytes for the lifetime of the infected individual. In the developed world half of all children become infected within the first decade of life; in the developing world the number is close to 100%. This infection is normally kept in check by EBV-specific immune responses, especially by cytotoxic CD8+ T cells which eliminate proliferating and lytically infected B cells. 44 Cigarette smoking is another major risk factor for MS, which appears to accelerate disease progression. 45
Although the cause of T cell activation has not been firmly established, what is clear is that auto-reactive lymphocytes move from the bloodstream into CNS parenchyma across the blood-CNS barrier (BCNSB).40,41 This occurs through a sequential and coordinated process involving the binding of adhesion molecules with respective ligands, which leads to the capture of activated leukocytes on to the inside surface of blood vessels and their rolling along the endothelium until they become stationary and then anchored into position 46 (Fig. 1). The first stage of this process involves selectin molecules on the endothe-lium interacting with counter-ligands on the leukocyte surface. For binding to occur the counter ligand must be decorated with specific carbohydrate side-chains, such as 2,3-sialylated and a1,3-fucosylated core 2 decorated O-glycans carrying the sialyl Lewis X (sLex) motif as a capping group.47,48 Anchoring of the leukocytes occurs by an interaction beween integrin proteins (either α4β1 or α4β7 integrin) on the leukocytes and vascular cell adhesion molecule-1 (VCAM-1) or mucosal addressin cell adhesion molecule-1 expressed on the endothelial cell layer. The immune cells then move across the BCNSB through tiny spaces in the endothelium and mount an attack on host cells; normally they would target infectious agents.46,49
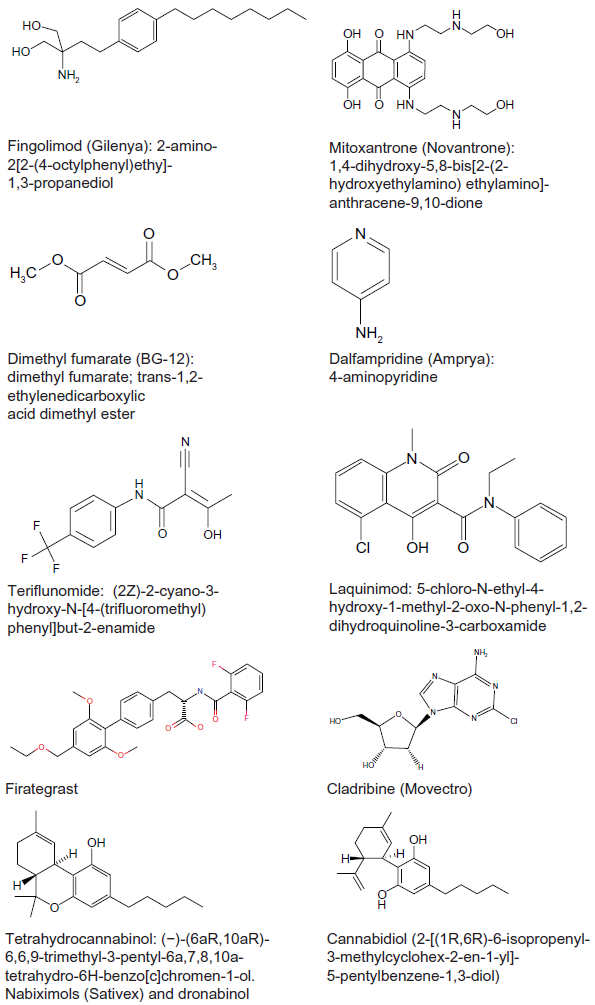
Small molecule drugs and drug candidates for the treatment of MS.
The clonal and oligoclonal expansion of lymphocytes in the CNS, particularly CD4+ T and CD8+ T cells, is amplified by pro-inflammatory cytokines through the recruitment of naive microglia and mediated by IFN-γ and IL-12.50,51 These lesions grow slowly by radial expansion as focal brain inflammation fades into diffuse parenchymal microglial activation resulting in extensive abnormalities in normal appearing white matter. 52 The resultant perivascular sclerotic plaques throughout CNS white matter, which are the pathological hallmark of MS, leads to breakdown of the BSNSB, partly through the action of interleukins 17 and 22. BCNSB disruption permits the movement of more leukocytes into the CNS parenchyma where they probably contribute to the process of neurodegeneration.2,53,54
Although grey matter lesions have been known to occur in MS for some time, 55 they have attracted much less interest than white matter lesions. This is largely because of difficulties associated with visualizing cortical grey matter lesions using conventional histochemical staining procedures. Improvements in MRI have brought grey matter changes to the fore and the importance of such change is emphasised by the demonstration that long-term disability correlates with the loss of grey matter, but not white matter loss. 56 Further support derives from a post-mortem study showing that the loss of corticospinal axons is the major contributor to the disability associated with both ppMS and spMS. 57
Assessment of symptoms, Disease Progression and the Efficacy of Drug candidates
Clear criteria have been established for the diagnosis of MS, which have recently been revised and simplified, with greater emphasis now placed on imaging to demonstrate the dissemination of CNS lesions over both space and time. 58 These criteria allow MS to be distinguished from other neurological disorders and CIS.
The main clinical measure of disability progression is the Expanded Disability Status Scale (EDSS), which was first developed in 1955, as the Disability Status Scale, a 10-point scale of disease severity ranging from 0 (no disability) to 10 (death from MS) and subsequently refined to create the 20-grade EDSS by dividing each grade into two to increase its sensitivity. 59 In the low range (0-3.5) it reflects a modest-to-moderate change in one or more of the functional systems: vision, brainstem, pyramidal, cerebellar, sensory, bowel and bladder, cerebral and ambulation. A score above 4.0 primarily reflects a dysfunction of gait, from 6.0-7.5 exclusively reflects walking ability and a score of 8.0 marks a complete loss of ambulation.
The EDSS is the only scale recognized by regulatory agencies, even though it has a number of shortcomings, including poor reproducibility, a failure to adequately assess upper limb function (with its reliance upon ambulation) and insensitivity to cognitive decline.60,61 In addition, the sensitivity of the EDSS to detect change in the clinical state of people with MS (pwMS) is poor and seems weakest at the lowest and highest ranges of the scale. In response to these limitations and concerns, the National Multiple Sclerosis Society's Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis developed the MS functional composite (MSFC). 62 It encompasses objective quantitative tests of ambulation (a timed 25-foot walk), arm function (9-Hole Peg Test) and cognitive function (a paced auditory serial addition test). Change in MSFC over the first year of observation is predictive of subsequent change in the EDSS, which suggests that the MSFC is a more robust and sensitive measure than the EDSS. It also displays a superior correlation with MRI variables, including brain atrophy, and with pwMS-reported disease-related quality of life. Also, short-term change in MSFC correlates with future clinical and MRI status.62,63 Even so, change in EDSS persists as the standard measure of disease progression in clinical trials.
Assessing disease progression in MS is further complicated by the absence of a consistent definition of data needed to claim that a drug candidate meaningfully impacts disease progression. A 1.0 step increase for individuals with an overall EDSS of 6.0 confirmed at 3 months is most commonly used. A recent longitudinal follow up of pwMS in a phase III trial indicated that worsening on EDSS of 1.0 point confirmed at 6 months is predictive of clinically significant disability at 8 years. 64 Nonetheless, despite being the gold-standard measure of MS disease progression, its validity remains uncertain and the optimal change that predicts long-term permanent sustained disability remains undefined.65,66 MSFC provides a more precise and responsive measure of change and so has substantial advantages over EDSS in MS clinical trials.62,67,68 The most recent use of the MSFC was the phase III trial of dalfampridine where the primary outcome measure was performance on a timed 25 foot walk compared with baseline.69,70
The most common primary outcome measure in pivotal MS clinical trials is relapse rate. A relapse is defined as new or worsening symptoms that last 24 hours in duration and occurs in the absence of fever or infection. Within a clinical trial, relapses can be identified by notification from pwMS, usually at a scheduled study visit and usually quantified as: (1) annualized relapse rate (ARR); (2) the average number of relapses per patient; (3) proportion of relapse-free pwMS; (4) proportion of pwMS relapsing; and (5) the cumulative probability of relapse. 71 However, the utility of using measures of relapses in clinical trials has been questioned on the basis that there is no clear link between relapse and long-term disability. 72 In addition, the number of relapses declines with time people with rrMS and are absent in both ppMS and spMS. 73 Such concerns have led to an increased use of MRI, which has now become the accepted surrogate primary outcome measure in proof-of-concept placebo-controlled clinical trials of new immunomodulatory drug candidates for the treatment of rrMS. 74
There are two types of MRI scan: T1-weighted and the T2-weighted images, which distinguish fat from water differently. In T1-weighted images water is darker and fat brighter, whereas the opposite is the case in T2-weighted scans. Since myelin is predominantly lipid (and thus hydrophobic), areas of demyelination hold more water and so show up as either a bright white spot or a darkened area depending on the type of scan used: T2- and T1-weighted scans, respectively. Contrast agents can be used to enhance the sensitivity of the T1-weighted MRI scans. The most commonly used contrast agent is gadolinium diethylenetriamine-pentaacetic acid (Gd-DTPA) and is principally used to visualise the number of new plaques in the brains of people with MS, but also to assess breakdown of the BCNSB. 75 This measure helps distinguish between acute (or active) plaques and chronic (or non-active) lesions. In the brain, MS plaques are commonly round or ovoid and range from a few mm to more than 1 cm in size. They are often found in the brainstem, cerebellum, and periventricular white matter. 76
Although the relationship between the clinical and subclinical changes on MRI and long-term disability in pwMS remains unclear, a combined measure of 1-year changes in MRI lesions and relapses after IFNβ therapy successfully predicted the subsequent effect on 2-year EDSS worsening. This short-term combined measure appears to be a surrogate for disability progression over a longer term when evaluating the effect of IFNβ-1a in rrMS. 77
Grey matter demyelination can be very extensive in MS, especially in the chronic phase of the disease. 52 Focal cortical lesions are difficult to visualise using conventional MRI scans because they are small and have poor contrast with the surrounding normal grey matter; partial volume effects from the CSF also confounds their detection. However, the sensitivity of MRI has been improved by the use of double inversion recovery sequences and thus cortical lesions can now be visualised in the intact brain. They have been seen in all the major clinical phenotypes of MS, including CIS. Brain atrophy provides another measure of neuronal loss. The progressive loss of brain volume is an important feature in the pathophysiology of MS and is reflected by widening of the inner and outer CSF compartments, especially in the progressive stage of the disease. 76
In addition to EDSS and MSFC, a number of other clinical outcome measures have been proposed for use in the study of MS. These include the Multiple Sclerosis Quality of Life, the Multiple Sclerosis Quality of Life Inventory, the Functional Assessment of Multiple Sclerosis, the Health-Related Quality of Life Questionnaire for Multiple Sclerosis, the Scripps Neurological Rating Scale and the Medical Outcomes 36-item Short-Form Health Survey.61,68 An increasingly popular scale is the Multiple Sclerosis Impact Scale (MSIS-29). This scale was developed to complement existing tests and provide a disease-specific measure of Health-Related Quality of Life that uses the perspective of pwMS on disease impact in MS. It aims to capture outcomes relevant to pwMS that are sometimes overlooked by clinicians. 78 There are also tests to assess specific symptoms, such as the Modified Fatigue Impact Scale 79 and the Multiple Sclerosis Spasticity Scale. 80
Current Ms Medicines
Immunomodulatory drugs
Table 1 summarizes the drugs approved by the FDA for the treatment of MS. It indicates that MS pharmacotherapy is dominated by immunomodulatory drugs, which were developed on the basis that MS is an autoimmune disease. This hypothesis posits that T-lymphocytes specific for myelin antigens initiate an inflammatory reaction in the CNS, which ultimately leads to demyelination and subsequent neuronal loss. Supporting evidence is derived primarily from studies on a single animal model, EAE. The origins of EAE date back to the 1920s, with studies inducing spinal cord inflammation in rabbits by inoculation with human spinal cord. Since then, EAE has been induced in many different species, including rodents and primates. 81 EAE reproduces many of the clinical, neuropathological and immunological aspects of MS, so it is a model that has good construct validity. 82 However, its predictive validity has been questioned because the therapeutic efficacy of drug candidates in the model have not always been translated into efficacy in pwMS.81,83,84
FDA approved drugs for the treatment of MS.
Immunomodulatory drugs currently used for the treatment of MS are described in Table 2. The first immunomodulatory drugs approved were β-interferons (IFNβs). IFNs are a family of proteins, first identified in 1957, that have antiviral, anti-proliferative and immunomodulatory efficacy through mechanisms that are not yet completely clear. 85 IFNβ drugs have dominated the MS drug market for nearly two decades. The first to gain FDA regulatory approval was Betaseron (1993), followed by Avonex (1996), Rebif (2002) and Extavia (2009). These recombinant proteins are produced by expression in either Chinese hamster ovary cells (IFNβ-1a; Avonex and Rebif) or in E. coli (IFNβ-1b; Betaseron and Extavia). IFNβ-1a (but not IFNβ-1b) is glycosylated with a carbohydrate very similar to that found in human IFNβ and IFNβ-1b (but not IFNβ-1a) has an amino acid substitution at position 17 (serine replacing cysteine), which reduces the probability of erroneous disulfide bond formation. 2 Table 3 describes trials that directly compared one IFNβ drug with another in studies of people with rrMS lasting from 16 to 24 months.
Current immunomodulatory drugs for the treatment of MS.
The effect of IFNβ drugs on relapse- and disease-related outcomes in clinical trials of people with MS.
Weighted mean difference, random effects model. Significant differences between groups indicated by asterisk
P < 0.05;
P < 0.005;
P < 0.0001.
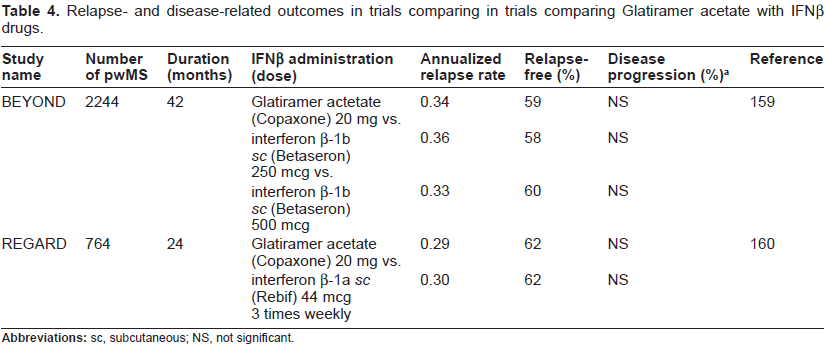
Another immunomodulatory therapeutic, which gained FDA approval for the treatment of rrMS in 1997, is glatiramer acetate (Copaxone). It is a random polymer of four amino acids (L-glutamic acid, L-lysine, L-alanine, and L-tyrosine) found in MBP, which is found in myelin. GA's mechanism of action is not known, but it may work as a decoy for the immune system.86,87 Systematic reviews of clinical trial data for glatiramer acetate in MS, indicates that the drug has partial efficacy in the treatment of rrMS in terms of relapse-related clinical outcomes, but is without any significant effect on clinical progression of disease, measured as sustained disability. Glatiramer acetate was also not effective in slowing the progression of disability in ppMS.88,89 The potential efficacy of glatiramer acetate in delaying the conversion of CIS to clinically definite MS has not yet been thoroughly tested. 90 Table 4 describes studies that directly compare glatiramer acetate to IFNβ drugs and indicate that it is equivalent in terms of both relapse and progression outcomes. However, its efficacy in reducing relapse rate is delayed by about 6 months. Glatiramer acetate administration, subcutaneously on a daily basis, is associated with flushing, chest tightness, sweating, palpitations, anxiety and local injectionsite; reactions; however, no major adverse effects were evident. 88
Relapse- and disease-related outcomes in trials comparing in trials comparing Glatiramer acetate with IFNP drugs.
IFNβ drugs have been available as first-line therapy for rrMS for nearly two decades now. They reduce relapse rate by about a third and make relapses milder. 92 However, they produce neutralising antibodies in about one third of those treated with an IFNP drug, which limits their efficacy. 93 IFNβ drugs are administered by either intramuscularly (Avonex) or subcutaneously (Betaseron, Rebif and Extavia), glatiramer acetate is given via the subcutaneous route. Injection of all IFNβ drugs and glatiramer acetate is often associated with side effects, principally injection-site reactions, moderate to severe flulike symptoms and the potential for liver damage. The skin reactions can be severe and lead to necrosis and lipoatrophy, which pwMS may experience after years of treatment. These side effects can be burdensome and lead to poor adherence and thus treatment failure. Injection-site reactions occur most often following subcutaneous administration (90% for those treated); these reactions occur in up to 33% of those using an intramuscular formulation.94,95 Glatiramer acetate is also associated with an immediate post-injection systemic reaction that can occur seconds to minutes after injection, lasting 10-20 minutes; it has not been determined to be dangerous to pwMS. 91 However, glatiramer acetate has a lower tendency to cause problems with skin, blood and liver than IFNβ drugs.
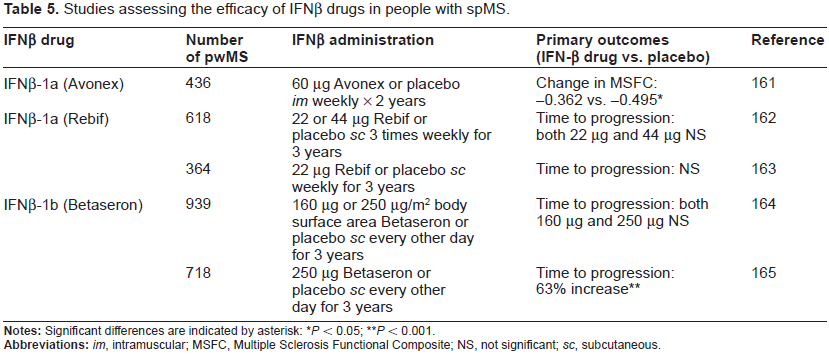
In addition, the efficacy of IFNβ drugs is variable, with 10%-50% of rrMS patients failing to respond to treatment. 96 A systematic analysis of the efficacy of IFNβ drugs on exacerbations and disease progression in rrMS showed only a modest effect after one and two years of treatment. 97 This conclusion is supported by long-term behavioural data over a 16-year follow-up period which showed no effect of IFNβ on long-term disability. 98 IFNβ treatment was also not associated with reduced disability progression in people with ppMS. 99 In addition, a systematic review of five randomised controlled trials of people with spMS (which includes most of the data shown in Table 5) that met pre-defined inclusion criteria, which included 1823 individuals taking IFNβ drugs and 1293 taking placebo, established that such treatment does not prevent the development of permanent physical disability. 100 A similar analysis of randomised controlled trials examining the efficacy of IFNβ drugs in the treatment of ppMS indicated that in the two studies that met the inclusion criteria (123 people with spMS), treatment was not associated with reduced disability progression. 101 However, a systematic analysis of three randomised controlled trials that met the inclusion criteria (639 treatment, 521 placebo) indicated that IFNβ treatment prevented the conversion from CIS to clinically definite MS, over two years of follow-up. 90
Studies assessing the efficacy of IFNβ drugs in people with spMS.
P < 0.05;
P < 0.001.
Overall, the data described above support the use of IFNβ drugs and glatiramer acetate to reduce relapse rates in MS, but they displayed little impact on disability-related outcomes. Even so, there is evidence to indicate that IFNβ drugs moderately reduce relapse rates, particularly in those with more active disease. 102 A systematic review of studies assessing the clinical efficacy and safety of glatiramer acetate in both rrMS and spMS indicated that it showed no beneficial effects on disease progression in both MS forms, a slight beneficial effect in the reduction of risk of relapses in people with rrMS and no benefit in those with progressive MS. 88
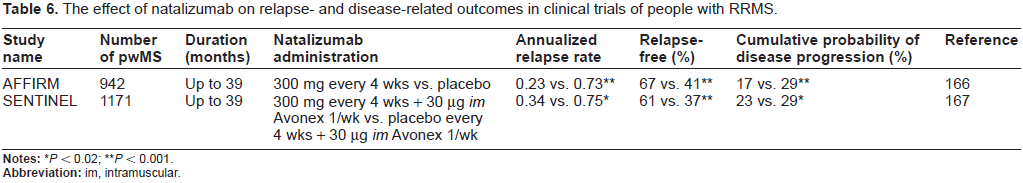
A more recent immunomodulatory drug is Tysabri (from Biogen Idec and Elan), which contains natalizumab–-a humanized monoclonal antibody (mAb) to α4:β1-integrin. This integrin exists on leukocytes and binds to cell adhesion molecules (vascular cell adhesion molecule-1 or mucosal addressin cell adhesion molecule-1) expressed on the endothelial cell layer. This interaction anchors immune cells to the endothelium to permit their movement across the BCNSB through tiny spaces in the endothelium; a process called diapedesis. 103 Under normal circumsances these cells then mount an attack on infectious agents within the CNS but, in the case of MS, they attack host cells.46,49 Thus, by inhibiting the interaction between α4-β1-integrin and cell adhesion molecules on the endothelium, natalizumab blocks the endothelial transmigration of lymphocytes into the CNS. 104 Two phase III clinical trials assessing relapse- and disease-related outcomes were pivotal in the approval of natalizumab for use in rrMS (Table 6). In a recent systematic review of available data that encompassed 3 studies and a total of 2,223 people with rrMS, natalizumab was found to reduce the number of participants who experienced relapses, the number of individuals who clinically deteriorated and showed increased MRI lesion activity at 2 years. 105
The effect of natalizumab on relapse- and disease-related outcomes in clinical trials of people with RRMS.
P < 0.02;
P < 0.001.
Natalizumab gained FDA approval in 2004 as first-line treatment of pwMS with highly active rrMS and for second-line treatment for pwMS failing to respond to IFNβ drugs or glatiramer acetate. However, its human use was suspended in 2005 because of two reports of progressive multifocal leukoencephalopathy (PML), including one fatal case; a third patient developed PML in another open label trial involving the use of natalizumab in Crohn's disease. PML is a rare, and usually fatal, viral disease caused by reactivation of the JC virus and characterized by progressive damage white matter. Natalizumab was re-introduced in the United States, with a black-box warning of PML, and approved in the European Union in 2006 after no additional cases of PML were identified in previously treated patients. The risk of developing PML increases after two years of treatment and with the use of immunosuppressant drugs prior to receiving natalizumab. The incidence of PML in pwMS receiving natalizumab over a 24-36 months of therapy is estimated to be 1/1000. 106 The robust benefits of treatment with natalizumab in pwMS with active rrMS is considered to outweigh the associated risk, but the long-term benefits and risks of its use are not yet known. The risk of developing PML is substantially reduced by limiting treatment duration to two years, avoiding treatment to pwMS taking immunosuppressive drugs and clinical vigilance, including demonstration of the absence anti-JC virus antibodies in serum prior to the commencement of treatment.106–108 With these caveats, natalizumab is now established as a useful second-line treatment for rrMS. A phase IIIb clinical trial multicenter, randomized, double-blind, placebo-controlled study to assess the efficacy of natalizumab in approximately 856 people with spMS is underway (Clinical Trials.gov identifier: NCT01416181).
In a systematic review and meta-analysis of all double-blind, randomised, controlled trials of studies of people with rrMS, three studies met the inclusion criteria. These included one placebo-controlled trial (942 patients) and two add-on placebo-controlled trials [one plus glatiramer acetate (110 participants) and the other plus IFNβ-1a (1171 participants)]. Thus in an analysis comprising a total of 2223 participants, natalizumab, was found to display robust evidence in favour of a reduction in both relapses and disability at 2 years, along with diminished MRI disease activity. 105 The reduction in ARR resulting from treatment with natalizumab is about twice that seen with IFNβ drugs and glatiramer acetate (Table 2), which is supported by a head-to-head study to assess whether natalizumab is more effective than IFNβ-1a (sc 44 µg; Rebif) on the basis of both clinical and radiological findings in 84 people with rrMS. In both groups, the ARR in the first 12 months of treatment was lower than in the 12 months before therapy, but EDSS reduction was significantly different between the two groups in favor of natalizumab (P < 0.002). The number of contrast-enhancing lesions displayed a greater reduction in the natalizumab group (P < 0.01) in the second year of treatment. 109
In a longitudinal study of 64 people with rrMS, after 1 year EDSS decreased by 0.47 points (P < 0.05), but there was no significant change after either 2 or 3 years. ARR decreased by 82%, 69% and 77% after 1, 2 and 3 years, respectively (all P < 0.001). 110 Thus, natalizumab reduces ARR and stabilizes EDSS score. This conclusion is supported by a longitudinal study of 620 people with rrMS from the pivotal natalizumab study AFFIRM with baseline EDSS scores of 2 or more. Cumulative probabilities of neurological improvement, defined as a 1.0-point decrease in EDSS score sustained for a minimum of 12 weeks were determined. Natalizumab increased the cumulative probability of improvement over 2 years by 69% versus placebo (P < 0.01). Sensitivity analyses indicated that natalizumab treatment conferred consistent benefits, including sustained EDSS improvement. 111
All of the medicines for MS discussed so far are large molecules. There are also small molecule drugs approved for the treatment for MS. The first of these is mitoxantrone (Novantrone), from Merck Serono. This immunosuppressive drug is a cytotoxic agent of the anthracenedione family that acts by intercalating with DNA and inhibiting topoisomerase. Because mitoxantrone inhibits T-cell, B-cell, and macrophage proliferation, it was developed for MS and, in 2000, gained FDA approval for use in the reduction of neurological disability and/or frequency of clinical relapses in pwMS with spMS, prMS, or worsening rrMS. 112
A systematic review compared mitoxantrone to placebo using data from 4 trials 113 and a second review included the same 4 trials as well as preliminary and unpublished data from an on-going study. 114 Among the 4 trials included in both reviews, there was some heterogeneity among the types of MS, the dose of mitoxantrone used, and study duration. Nonetheless, mitoxantrone was found to be moderately effective in reducing both disease progression and the frequency of relapses in all subgroups, at least in the short-term. However, because of safety concerns (principally the long-term risk of therapy-related infertility, leukaemia and cardiotoxicity), the use of mitoxantrone in MS requires careful selection of pwMS, drug administration, and drug monitoring, with the lifetime cumulative dose strictly limited to 140 mg/m2, or 2 to 3 years of therapy.112,113 As a result of these safety concerns, it is considered a second-line therapeutic agent and used cautiously.
The second small molecule MS medicine is fingolimod (Gilenya; Gilenia in Europe), which was developed by Novartis. Fingolimod is a structural analog of intracellular sphingosine that is phosphorylated by sphingosine kinase 2 in vivo, and exerts its effects by mimicking sphingosine 1-phosphate (S1P) and binding to four of the five S1P receptors on lymphocytes. This binding leads to internalization of activated S1P receptors, and their down-regulation. In the absence of S1P receptor signalling, CD4+ and CD8+ T cells and B cells are unable to egress from secondary lymphoid tissue, which reduces the number of lymphocytes in the blood by about 70%. 115 This sequestration of activated leukocytes within the lymphoid tissue interrupts their movement into the CNS.
Clinically, fingolimod reduced relapse rate, MRI lesions, brain-lesion activity and loss of brain volume as measured by MRI in comparisons with both placebo and IFNβ-1a (Avonex; Table 7). On this basis it gained regulatory approval for the treatment of rrMS, by the FDA in 2010 and the European Medicines Agency (EMA) the following year. It is indicated in the US as first-line treatment for rrMS, at a recommended dose of 0.5 mg once daily, to reduce the frequency of clinical exacerbations and delay the accumulation of physical disability. In the EU, fingolimod is indicated for the treatment of people with highly active rrMS despite treatment with IFNβ drugs/glatiramer acetate, or pwMS with rapidly-evolving severe rrMS.
The effect of fingolimod on annualised relapse rates in people with rrMS.
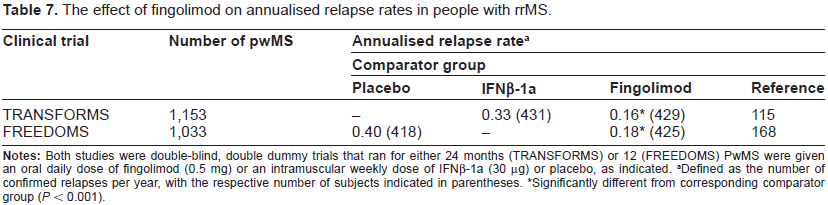
Defined as the number of confirmed relapses per year, with the respective number of subjects indicated in parentheses.
Significantly different from corresponding comparator group (P < 0.001).
Fingolimod reduced the annualised relapse rate relative to both placebo and IFNβ-1a, and in one of the Phase III trials (FREEDOMS, but not TRANSFORMS) it also reduced the risk of disability progression (Table 7). Adverse events include initial and transient bradycardia, transient liver dysfunction, macula edema and a mild reduction in measures of respiration. In addition, the TRANSFORMS study was associated with two deaths, which occurred as a consequence of disseminated Varicella zoster infection and Herpes simplex encephalitis. Serious safety concerns have recently emerged with reports of 11 deaths among pwMS who took the drug; seven are unexplained, three were caused by heart attack and one was caused by a disruption to heart rhythm. Both the FDA and the EMA have initiated investigations of these serious adverse events.
People with rrMS who received fingolimod in the TRANSFORMS study remained on their original dose (0.5 or 1.25 mg); those who received IFNβ-1a (Avonex) were randomized to receive either 0.5 or 1.25 mg fingolimod. Fingolimod-treated subjects experienced a consistently low annual relapse rate after both 1 and 2 years of treatment. They also displayed a reduction in relapses and MRI brain lesions over 2 years compared to pwMS switched to fingolimod. In those who were switched to fingolimod, the annual relapse rate in year 2 was reduced by 31%, and the number of new or newly enlarged T2 lesions in the brain was reduced by 67%. 116 In addition, a meta-analysis comparing the ARR of people with rrMS treated with fingolimod versus those taking glatiramer acetate or IFNβ drugs, using evidence from both placebo-controlled and head-to-head studies, indicated that fingolimod displayed superior efficacy.117
All of the drugs described above are immunodulatory agents that aim to modify the course of MS by slowing the progression of disability in a clinically meaningful way. In addition to this approach, is the strategy of pharmacotherapy to improve specific symptoms of MS. There are two examples of such compounds that have gained regulatory approval, both of which are small molecules.
Symptomatic drugs
The first of these is Ampyra (dalfampridine extended release tablets) from Acorda Therapeutics. It has been approved by both the FDA and EMA for the treatment of MS. Dalfampridine is the broad spectrum potassium channel blocker 4-aminopyridine, which works by extending the action potential of presynaptic neurones. This leads to improved impulse flow along axons and increased release of neurotransmitter (mainly acetycholine) at the neuromuscular junction, resulting in an improvement in motor function. Its approval was on the basis data from the two phase III clinical trials that demonstrated that Ampyra (10 mg twice daily) improved walking speed as measured by the timed 25-foot walk. by an average of 25%. Though modest, this improvement was associated with a reduction in ambulatory disability in pwMS.69,70,118 However, only one-third of the pwMS who received the drug were consistent responders. The safety profile of dalfampridine is of concern as potassium channels are intrinsic to normal function, particularly of excitable tissues such as the heart and CNS. It is, therefore, not surprising that dalfampridine has a narrow therapeutic range, with adverse events mainly related to stimulatory effects on the nervous system. The most common reported serious adverse events were MS relapse and epileptic seizures. 69 However, an analysis of multiple published studies suggest that adverse events are dose-related, mild to moderate and transient. 119
The second symptomatic treatment is nabiximols (Sativex), from GW Pharmaceuticals, which was recently granted regulatory approval but not by the FDA. This cannabis-based oral spray contains a defined quantity of specific cannabinoids, particularly tetrahydrocannabinol and cannabidiol, which are cannabinoid CB1 and CB2 receptor agonists, respectively. It has regulatory approval in Canada and a number of European countries on the basis of clinically relevant improvement in spasticity in people with MS. 120 Adverse events include somnolence, nausea and dizziness.
Emerging MS Medicines
A number of potential MS drugs are currently in clinical trials and most of those at Phase II or III are summarised in Table 8. These include both large and small molecules. The large molecules are mainly humanized mAbs, including alemtuzumab, daclizumab and ocrelizumab, but also include other types of molecules, such as tovaxin, firategrast, dimethyl fumarate, teriflunomide and dronabinol.
Drug candidates in phase II or III clinical development for MS.
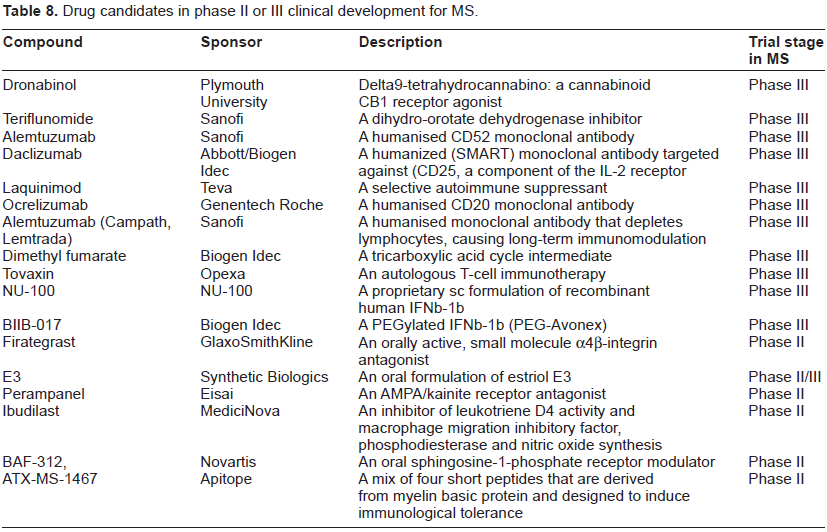
Alemtuzumab (Campath, Lemtrada) is a humanised monoclonal antibody that is currently approved by the FDA for the treatment of B-cell chronic lymphocytic leukemia. Its target is CD52, a 12 amino acid glycoprotein expressed throughout the immune system on T and B lymphocytes, natural killer (NK) cells, dendritic cells, and most monocytes. The exact function of CD52 in immune cells is not known, but CD52 cross-linking has been shown to trigger human T cell activation 121 and alemtuzumab depletes CD4+ and CD8+ lymphocytes, causing long-term immunomodulation. A 36-month randomized, blinded 3 year Phase II trial in therapy-naïve individuals with early rrMS (EDSS of 3 or less) compared alemtuzumab treatment with that of IFNβ-1a (Rebif). They received either subcutaneous IFNβ-1a (at a dose of 44 mg three times per week) or annual intravenous cycles of alemtuzumab (at a dose of either 12 mg or 24 mg per day) for 36 months. Both doses of alemtuzumab reduced accumulated disability at 6 months, caused fewer relapses than those receiving IFNβ-1a and produced significant benefits with respect to comparator for early MRI lesion changes and brain volume. In a 5-year follow-up study comparing alemtuzumab with IFNβ-1a in early, active rrMS, alemtuzumab remained significantly more efficacious than IFNβ-1a. 122 However, alemtuzumab was associated with serious adverse events, with three treated individuals developing immune thrombocytopenic purpura, one of whom died. Other adverse events, as compared with the IFNβ group, included thyroid disorders (23% vs. 3%) and infections (66% vs. 47%), along with elevations in liver function tests. 123 In a subsequent two year, phase III clinical trial of 322 people with rrMS, alemtuzumab reduced relapse rates by 55% compared to IFNβ-1a (Rebif), but there was no difference in accumulated disability (measured by EDSS) between the two groups. 124 Alemtuzumab treatment in people with rrMS does increase the risk for the development of autoimmune disease, but this risk is time-limited and modified by external factors. 125 Nonetheless, it carries a black box warning of serious hematological toxicity, infusion reactions, and opportunistic infections.
Daclizumab (from Biogen Idec) is widely used to prevent rejection after allogeneic tissue transplantation. It is a humanized mAb that binds to the alpha subunit of CD25, the high affinity IL-2 receptor. CD25 expression is elevated in activated T cells and daclizumab selectively inhibits the binding of IL-2 to CD25, and thus blocks T-cell activation. 126 Unlike alemtuzumab, treatment with daclizumab does not cause an immediate deletion of CD25-bearing cells. A multinational, double-blind, placebo-controlled phase IIb trial (SELECT) of sc daclizumab (300 mg/month or 150 mg/month, or placebo) in 600 people with rrMS showed reduced ARR (54% and 50% in in the low and high groups, respectively). There was also evidence to indicate that daclizumab improved quality of life and slowed disability progression, as measured by the MS impact scale (MSIS-29) and EDSS, respectively.127,128 Two phase II extensions of the SELECT study are underway, along with a Phase III trial. A recent systematic analysis of the use of daclizumab (both alone and combined with other treatments) in the treatment of rrMS concluded that, in general, it is effective and safe. However, none of the studies met the inclusion criteria for this Cochrane analysis. 128
There is accumulating evidence suggesting the involvement of B lymphocytes in the pathophysiology of MS. Rituximab |(from Roche) is a genetically engineered chimeric murine/human mAb that targets the CD20 antigen, a transmembrane phosphoprotein expressed only by pre-B and mature B cells. It depletes B-lymphocytes via complement-dependent cell lysis and antibody-dependent cellular toxicity.
In a study of 104 people with rrMS, rituximab (iv 1000 mg on days 1 and 15) reduced the rate of relapse at 24 weeks but not at 48 weeks.129,130
Ocrelizumab is another anti-CD20 mAb that is under development by Genentech and Roche for the potential iv treatment of both rrMS and ppMS. A phase II study of people with rrMS showed that Ocrelizumab reduced the total number of Gd-enhancing lesions and T1-weighted MRI. 131 Phase III trials for both rrMS and ppMS are on-going. This includes a randomized, double-blind, parallel-assignment, safety and efficacy trial (OPERA) to evaluate ocrelizumab compared with IFNβ-1a (Rebif) in people with rrMS (Genentech Inc, Press Release October 20, 2011) and a multinational, placebo-controlled phase III trial (ORATORIO) in people with ppMS (Genentech Inc, Press Release October 20, 2011).
Tovaxin is being developed by Opexa, under license from the Baylor College of Medicine, for the potential sc treatment of MS. It is an autologous T-cell immunotherapy that consists of in vitro expanded myelin-reactive T-cells, manufactured against up to six immunodominant peptides derived from three myelin antigens. A phase II study of people with rrMS, showed no statistically significant clinical or radiological benefit. 132 A pivotal, phase III study is planned (Opexa Therapeutics Inc Press Release April 15, 2011).
Small molecule drug candidates in clinical trials for MS are shown in Table 8 and some are described in more detail below.
Biogen Idec has submitted a New Drug Application to the FDA for the use of dimethyl fumarate (BG-12; the methyl ester of fumaric acid) for the treatment of rrMS. Fumaric acid esters are a group of low molecular weight compounds that have been used in the treatment of moderate to severe psoriasis (another chronic immune condition) since 1959. Long-term tolerability and safety are considered to be favourable, but there are several case reports describing acute kidney injury or Fanconi syndrome. 133 Fumarate is an intermediate in the tricarboxylic acid cycle and is formed by the oxidation of succinate by the enzyme succinate dehydrogenase. Fumarate is then converted by the enzyme fumarase to form malate. However, the mechanism by which therapeutic efficacy is achieved is not clear, although there is data to indicate that fumarate treatment induces IL-4-producing Th2 cells in vivo and generates type II dendritic cells that produce IL-10 instead of IL-12 and IL-23. This is consistent with Th1- and Th17-mediated autoimmune diseases such as psoriasis and MS. 134
Two randomized, placebo- and active-controlled, double-blind, parallel-group Phase III studies (DEFINE and CONFIRM) of dimethyl fumarate for rrMS are underway to assess the safety and efficacy of dimethyl fumarate compared with glatiramer acetate. Top-line results showed that 240 mg of dimethyl fumarate, administered either twice or three times a day demonstrated significant efficacy and favorable safety and tolerability profiles (Biogen Idec press releases, 2011). The most frequently reported adverse events across the 3 study groups were flushing, MS relapse, nasopharyngitis, headache, diarrhea, and fatigue. These events were more common with dimethyl fumarate than with placebo, with the highest incidence in the first 30 days of treatment and decreasing thereafter.
There is published report of a phase II 24 week trial of 257 people with rrMS who received oral fumarate 120 mg once daily, 120 or 240 mg three times daily, or placebo. The study demonstrated a 69% reduction in the mean number of Gd-enhanced lesions (the primary endpoint) in the 240 mg three times daily treatment group. This group also showed a reduced number of new or enlarging T2-hyperintense and T1-hypointense lesions and reduced relapse rates. 116
Teriflunomide, from Sanofi, is an active metabolite of the rheumatoid arthritis drug leflunomide. It inhibits dihydro-orotate dehydrogenase, a key enzymatic step required in pyrimidine synthesis. Because the production of activated T-cells largely depends on de novo pyrimidine synthesis, pyrimidine depletion is thought to result in the inhibition of immune cell proliferation.135,136 A US filing was submitted in 2011 and supported by data from five phase III studies: TERACLES, TENERE, TOWER, TOPIC and TEMSO. Top-line data indicated that teriflunomide was efficacious with no serious adverse events. For example, in the TEMSO trial, it significantly reduced ARR in people with rrMS compared to placebo at 2 years and was well tolerated at both doses administered (7 and 14 mg).
A 36-week, double-blind, Phase II study (enrolling people with rrMS and spMS) evaluated the effectiveness of two doses of teriflunomide (7 or 14 mg daily) versus placebo. Both doses of teriflunomide reduced the relapse rate by 31% over placebo (ARR of 0.37 versus 0.54, respectively). Both doses of teriflunomide also reduced the number of MRI-visualised lesions by >61%, including: fewer combined unique active lesions, T1 Gd-enhancing lesions, and new or enlarging T2 lesions. 137 In an interim analysis of a long-term open-label extension of this study, it was found that the most common adverse events were mild infections, fatigue, sensory disturbances and diarrhoea. In terms of efficacy, annualised relapse rates remained low, with minimal disability progression and several MRI parameters indicated an apparent pathological benefit. 138
Laquinimod is another novel oral agent for the treatment of MS. It is being developed by Teva, under license from Active Biotech, and is thought to act by shifting the immune response from Th1 to Th2. A phase III trial of its parent compound (linomide) was terminated early because of cases of myocardial infarction, pleuropericarditis, and other serious adverse events. 139 Laquinimod appears to have a superior efficacy and safety profile. 140
In a 24-week phase II trial, two doses of laquinimod (0.3 and 0.6 mg) were given daily to 306 people with rrMS. Both doses were well tolerated and the highest dose reduced the formation of active lesions visualized by MRI. 141 Two Phase III studies of people with rrMS (BRAVO and ALLEGRO) are underway.
BRAVO is designed to evaluate the safety and efficacy of the drug and to provide risk-benefit data comparing the drug to IFNβ-1a (Avonex). At 24 months, a significant reduction in ARR of 21% was achieved, along with a 33% reduction in the risk of disability progression (as measured by the EDSS), and a 28% reduction in the risk brain volume loss, as measured by MRI (Teva Pharmaceutical Industries Ltd, Press Release October 19, 2011).
The ALLEGRO trial was a 24-month randomized, placebo-controlled, double-blind study to evaluate the safety and efficacy of daily oral administration of 0.6 mg of the drug in 1106 people with rrMS. A modest reduction in the mean ARR compared with placebo (0.30 versus 0.39, respectively) was observed, along with a reduction in the risk of confirmed disability progression (11% versus 16%, respectively). In addition, the mean cumulative numbers of Gd-enhancing lesions and new or enlarging lesions on T2-weighted images were lower for pwMS who received laquinimod compared with those who received placebo; reductions of 27% and 58%, respectively 142 (Teva Pharmaceutical Industries Ltd, Press Release March 15, 2012).
Firategrast is being developed for the potential treatment of MS by GlaxoSmithKline, under license from Tanabei. It is an orally active, small molecule α4β-integrin antagonist with a shorter half-life than natalizumab that has demonstrated efficacy on imaging endpoints in a Phase II study of 343 individuals with rrMS. One of four treatments were administered twice a day: firategrast at a dose of 150 mg (49 individuals), 600 mg (95) or 900 mg (women) or 1200 mg (men) or placebo (99). A 49% reduction was observed in the cumulative number of new Gd-enhancing lesions at the highest dose tested. 143
NU100 is a proprietary recombinant human IFNβ-1b that is being developed by Nuron Biotech for the treatment of MS that is in a Phase III clinical trial involving 500 people with rrMS randomized to receive NU100, a marketed IFNβ-1b or placebo over a 12-month period.
BIIB017, from Biogen Idec, is PEGylated IFNβ-1a. It represents an extended-release version of Avonex and Rebif and so requires it be taken every 2-4 weeks, rather than 1 to 3 times weekly (Table 1). A phase III trial is underway to determine its efficacy in reducing ARR and MRI lesions at one year in people with rrMS in comparison to a placebo group.
Cladribine (Movectro), from Merck Serono, is a chlorinated purine analog (2-chlorodeoxyadenosine), and anti-neoplastic agent, that inhibits the enzyme adenosine deaminase, and thus interferes with the cell's ability to process DNA. In a Phase III trial of 1,326 pwMS that were randomly assigned one of two cumulative doses of cladribine (3.5 mg or 5.25 mg per kilogram of body weight), cladribine significantly reduced relapse rates, the risk of disability progression, and MRI measures of disease activity at 96 weeks. 144 However, there were serious safety concerns with this product, including, lymphocytopenia, Herpes zoster infections and neoplasms (including malignancies). 145 By the end of 2010, oral cladribine had been launched in Russia and Australia for rrMS, but in June 2011, Merck Serono withdrew the product from the market and withdrew their New Drug Application to the FDA, deciding that it was not commercially viable to conduct the additional clinical trials likely to be required for approval; their Marketing Authorization Application to the EMA was withdrawn in February 2011, following a final negative opinion from the EMA's Committee for Medicinal Products for Human Use.
Dronabinol is synthetic delta9-tetrahydrocannabinol (Δ 9 -THC). This cannabinoid CB1 receptor agonist is in a phase III clinical trial (CUPID) to assess its ability to slow the progression of disability in MS. The study is being carried out at Plymouth University in the UK and has recruited 493 participants who will each take part in the trial for 3 years; 3.5 years in some cases. 146
There are a number of drug candidates in development that aim to treat specific MS symptoms, including AbobotulinumtoxinA (Dysport; botulinum toxin type A-hemagglutinin complex). This protein produced by the bacterium Clostridium botulinum (botulinum toxin A) is currently in phase III clinical trials for the treatment of overactive bladder in people with MS who still void voluntarily. 147 It inhibits acetylcholine release and so is a neuromuscular blocking agent. Its acceptance for use in the treatment of muscle pain disorders is growing, with approvals pending in many European countries.
Concluding Remarks
MS is a chronic disease of the CNS that is characterised by progressive loss of neurologic function and is influenced by genetics, epigenetic and environmental factors. The immune system is central to its pathogenesis, with the disease probably initiated and perpetuated by self or foreign antigens. 148 Consequently, pharmacotherapy is dominated by immuno-modulatory drugs.
The market for such medicines has grown substantially over the last two decades, with the first three IFNβ drugs (Betaseron, Avonex and Rebif) and glatiramer acetate now well established as first-line, disease-modifying therapies. They generate handsome collective sales: $6.6 billion in 2009, 2 and according to Thomson Pharma Partnering Forecast, the market for MS drugs was $8.9 billion in 2008 and is predicted to rise to $15.6 billion in 2014. This is attributable to further growth in the sales of natalizumab and a more recent IFNβ-1b (Extavia), along with the emergence of new biologic immunomodulatory drugs (particularly mAbs) and small molecule immunomodulatory drugs (the first of which is fingolimod) that can be administered orally rather than by injection; a number of small molecule drug candidates are in clinical trials (Fig. 1 and Table 8). This is an important breakthrough as about half of pwMS receiving first-line immunomodulatory drugs discontinued use within 6 years. 149 This, together with evidence indicating that fingolimod has a superior efficacy profile to existing first-line biologic therapies, suggests that fingolimod has the potential to have a major impact on the treatment of MS.116,117
IFNβ drugs and glatiramer acetate have a long history as first-line treatments for rrMS and have been shown to reduce both clinical exacerbations and MRI lesion activity. The greatest benefit is seen with the commencement of treatment early in the course of the disease, particularly after the first demyelinating event (CIS). The choice of drug is determined by a number of factors, including patient preference, tolerability of the various side effects, the presence of antibodies to IFNβ drugs or glatiramer, clinical and MRI disease activity and disease course. Switching therapies is a sensible course of action for pwMS not responding to any of the first-line treatment options or those with very active disease. For these pwMS, treatment with natalizumab and fingolimod is recommended; fingolimod is, in fact, approved as first-line therapy in the USA.
A prerequisite of all MS drugs entering the market and remaining there is that the resultant clinical benefit (positive effects) outweighs the associated risk (potential harm). This metric has led to the temporary withdrawal of natalizumab (because of concern over the risk of PML) and the permanent withdrawal of cladribine from the market, along with the total cessation of its development. In addition, serious safety concerns associated with the use of fingolimod have led to the initiation of a reappraisal of the risk-benefit profile of this MS drug, and will probably lead to a requirement for more rigorous cardiovascular monitoring. Cost is another key influence on the current and future MS drug market since IFNβ drugs and glatiramer acetate are very expensive for the modest health gains they provide. 150 The patents for Betaseron, Avonex, Rebif and glatiramer acetate are due to expire soon, which opens the door for biosimilars drugs (eg, CinnoVex 151 ) to enter the market, probably from 2014 onwards. 152 This will erode existing sales, put downward pressure on price and impact future drug ranking and modalities of approval.
Another factor changing the market landscape, and thus providing more options for the treatment of MS, is the emergence of small molecule palliative medicines (such as dalfampridine and nabiximols) to treat specific symptoms of MS. However, the greatest potential for new blockbuster products comes with the prospect of new medicines that significantly impact disease progression, in all forms of MS. This is most likely to be achieved with neuroprotective agents. Evidence to support this hypothesis includes the following: (1) neuronal loss (but not loss of myelin) correlates with clinical symptoms;56,57 (2) in a16 year follow-up study of the pivotal IFNβ-1b trial in rrMS, neither MRI T2 burden change nor accumulation of new MRI lesions during relapses was predictive of disability or cognitive change; 153 (3)immunomodulatory therapieshave greatest efficacy when applied early in the disease course, have little effect on the long-term accumulation of disability and are essentially devoid of efficacy in the treatment of both spMS and ppMS (Table 5);2,3 and (4) the progression of disability in MS seems to occur either in the presence (the neuroinflammatory phase) or absence (the neurodegenerative phase) of focal inflammatory lesions. 154 Thus neurodegenerative change is a major driver of disability in MS, particularly in progressive phases of the disease, and so there is great potential for neuroprotective therapeutics to have a major impact on the treatment of MS. However, such medicines are not yet available. At present, there are no examples of a neurological disorder where a neuroprotective agent meaningfully slows the advance of neurologic disability.103,155 Indeed, this is the major challenge facing CNS medicines research. 170
Author Contributions
Analysed the data: AMP. Wrote the first draft of the manuscript: AMP. Contributed to the writing of the manuscript: AMP. Agree with manuscript results and conclusions: AMP. Developed the structure and arguments for the paper: AMP. Made critical revisions and approved final version: AMP. The author reviewed and approved of the final manuscript.
Competing Interests
The authors disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest. Provenance: the author was invited to submit this paper.